

Transition of D3c branch and novel recombination events contribute to the diversity of Coxsackievirus A6 in Beijing, China, from 2019 to 2023
- PMID: 40574752
- PMCID: PMC12202042
- DOI: 10.1093/ve/veaf036
Transition of D3c branch and novel recombination events contribute to the diversity of Coxsackievirus A6 in Beijing, China, from 2019 to 2023
Abstract
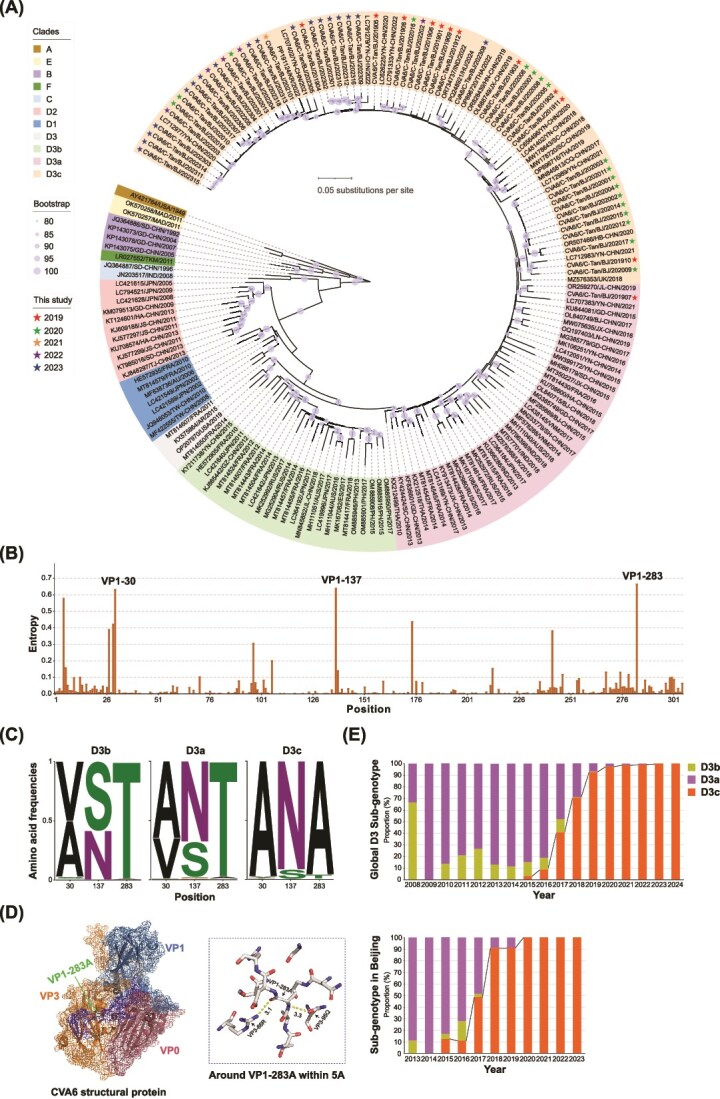
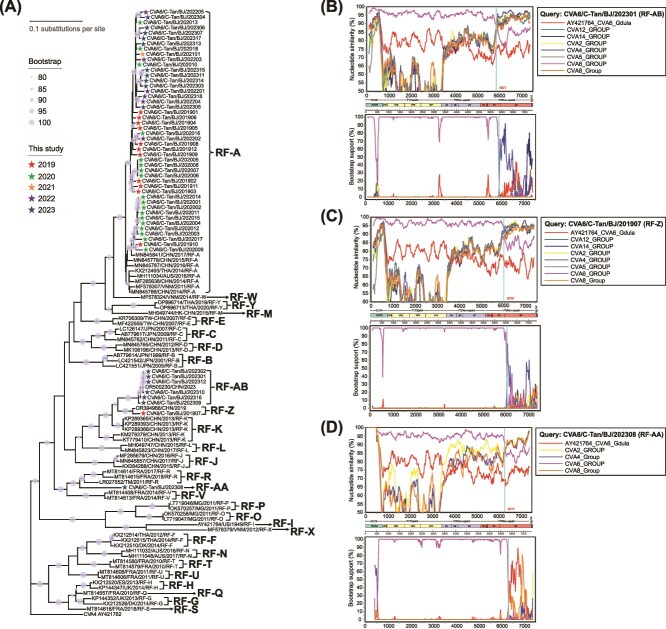
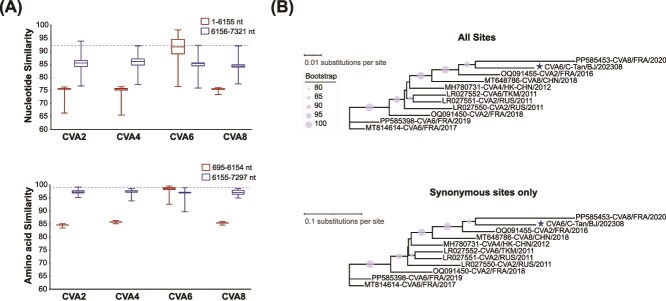
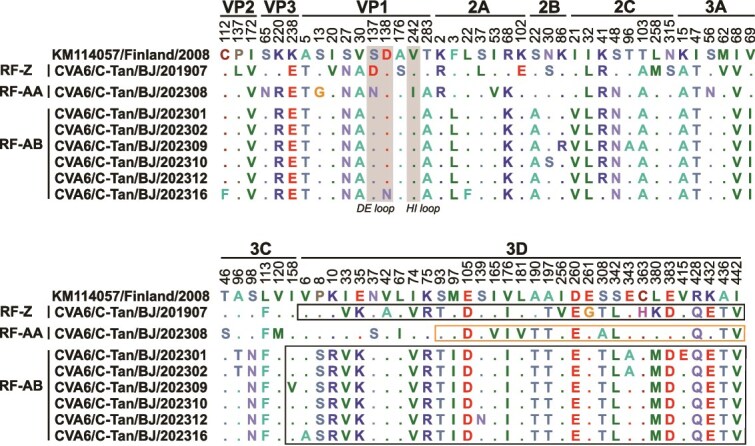
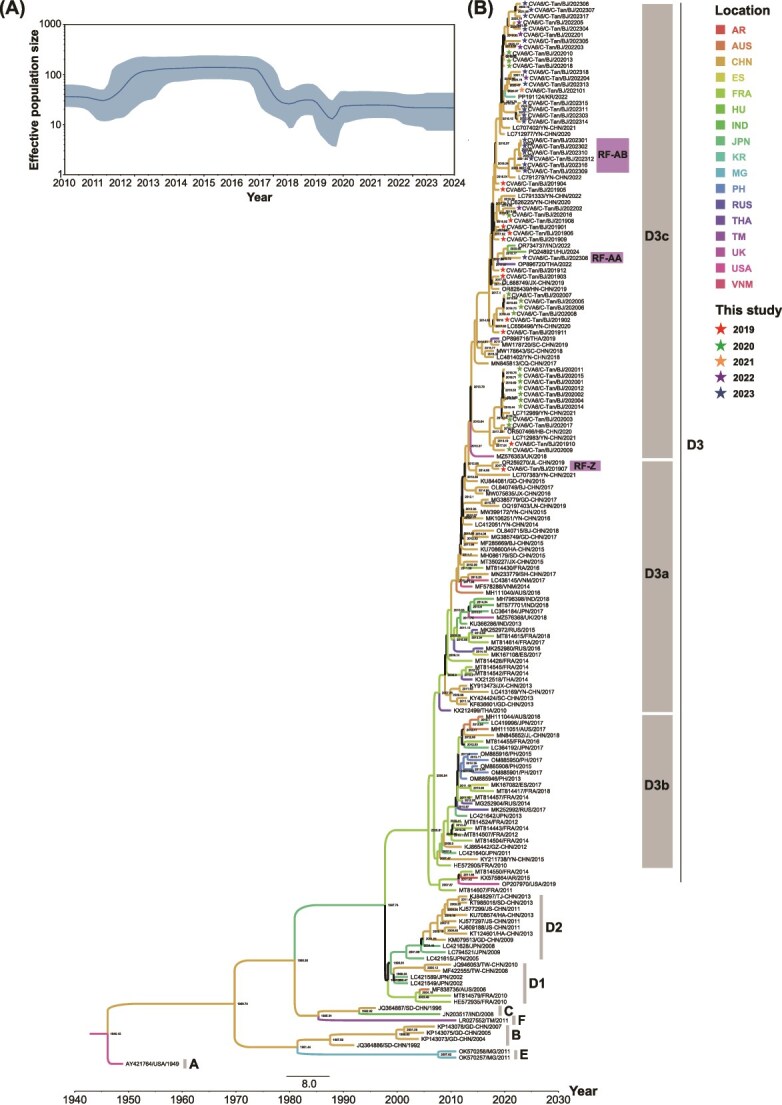
Coxsackievirus A6 (CVA6) is a major pathogen responsible for numerous outbreaks of hand, foot, and mouth disease (HFMD) worldwide. This study investigates the molecular evolution and recombination of CVA6 in Beijing, China. Full-length sequences of 54 CVA6 from Beijing (2019-2023) were obtained through metagenomic next-generation sequencing and Sanger sequencing. These sequences were compared with representative sequences from GenBank to analyse their phylogenetic characteristics, recombination diversity, and evolutionary dynamics. The 54 CVA6 strains co-circulated with those from multiple provinces in China, as well as from South Korea and Japan. Phylogenetic analysis revealed a novel D3c branch, with the VP1 T283A amino acid mutation identified as a key change in its formation. One sequence belonged to the D3a branch, while 53 sequences belonged to the D3c branch. Recombination analysis identified RF-A (46, 85.1%) and three novel recombinant forms (RFs): RF-Z (1, 1.9%), RF-AA (1, 1.9%), and RF-AB (6, 11.1%). Bayesian phylogenetic analysis estimated that the most recent common ancestor of D3c emerged in August 2013 (95% highest probability density (HPD): May 2012 to September 2014), with recombination events occurring in RF-Z (2017-2019), RF-AA (2019-2023), and RF-AB (2021-2023). In conclusion, we revealed a globally circulating CVA6 D3c branch and identified three novel RFs, providing valuable insights for the intervention and control of HFMD.
Keywords: Coxsackievirus A6 (CVA6); hand, foot, and mouth disease (HFMD); metagenomic next-generation sequencing; novel D3c branch; recombinant forms (RFs).
© The Author(s) 2025. Published by Oxford University Press.
Figures

References
Associated data
LinkOut - more resources
Full Text Sources
