

Glycosphingolipid synthesis is impaired in SLC35A2-CDG and improves with galactose supplementation
- PMID: 40576648
- PMCID: PMC12204976
- DOI: 10.1007/s00018-025-05759-w
Glycosphingolipid synthesis is impaired in SLC35A2-CDG and improves with galactose supplementation
Abstract
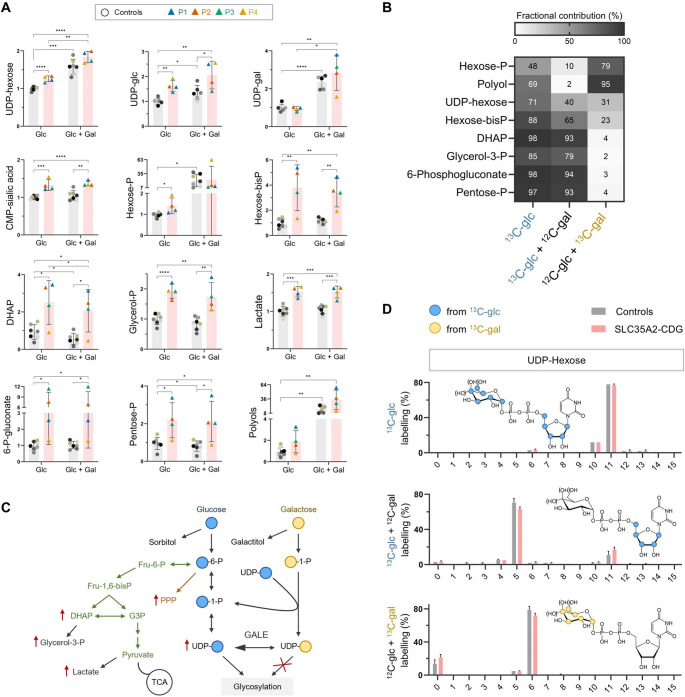
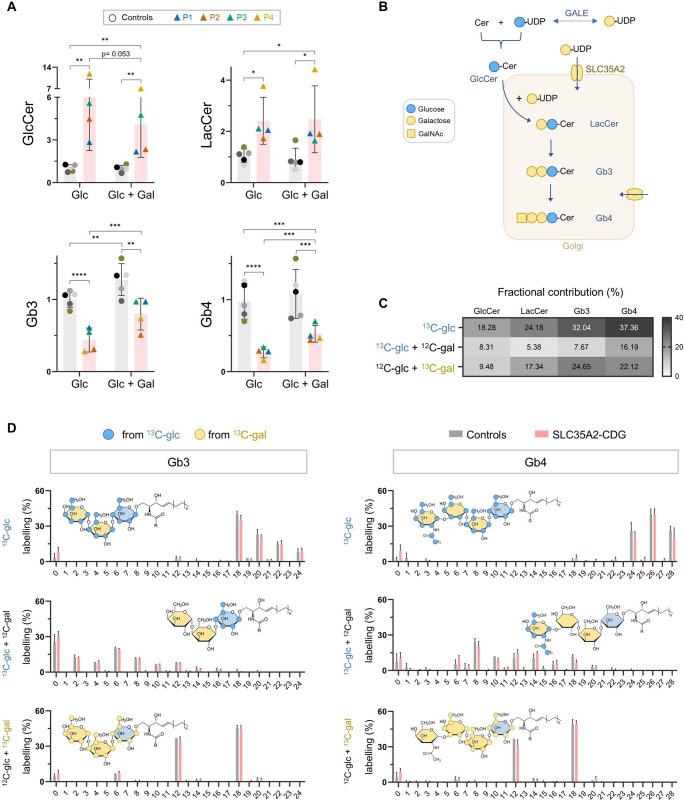
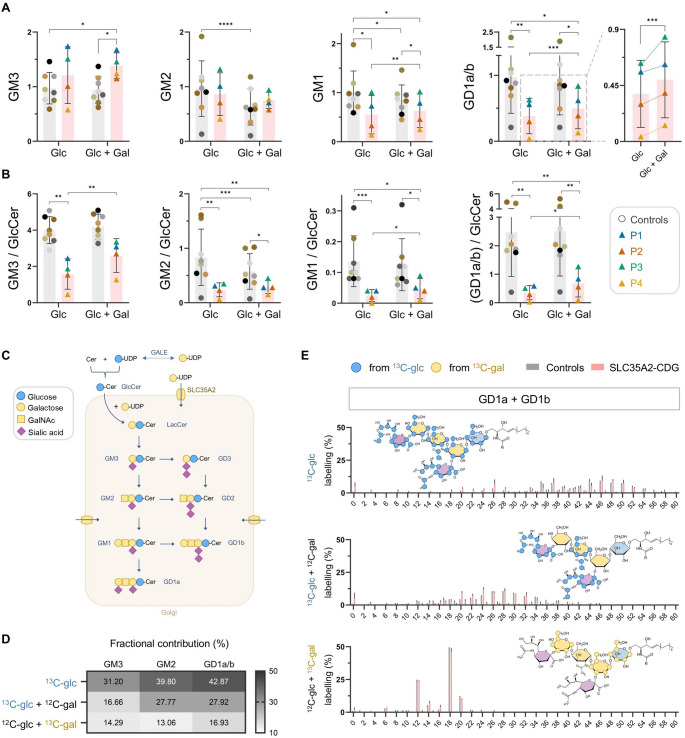
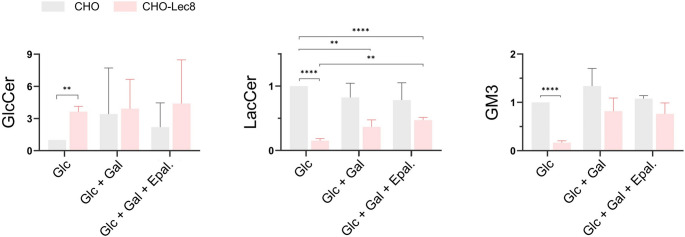
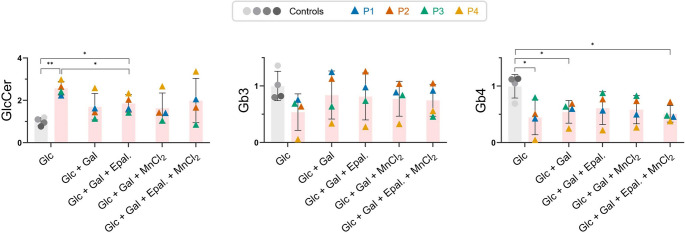
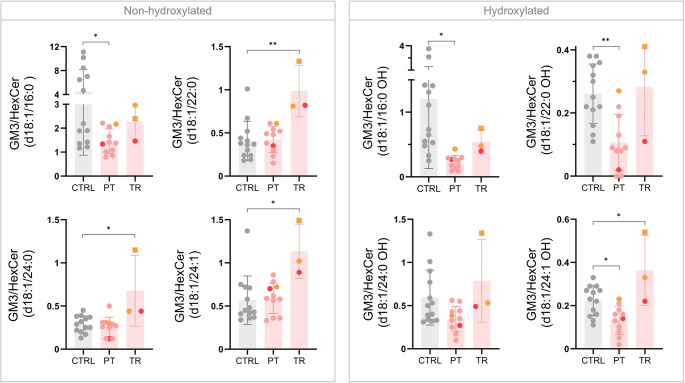
SLC35A2-CDG is an X-linked congenital disorder of glycosylation (CDG), characterized by defective UDP-galactose transport into the Golgi and endoplasmic reticulum and consequent insufficient galactosylation of glycans. Clinically, this translates into a range of predominantly neurological symptoms. Although the pathomechanism of this disorder is not fully understood, oral galactose supplementation has led to clinical and biochemical improvement in some patients. Here, we show that protein glycosylation (N- and O-linked) was only minimally disturbed in SLC35A2-CDG patient-derived fibroblasts. However, lipid glycosylation was significantly impaired, with accumulation of glucosylceramide and deficiency of digalactosylated glycosphingolipids (GSLs) and complex gangliosides. Galactose supplementation increased UDP-galactose, its transport into the Golgi, and improved deficient GSL synthesis through direct incorporation of the provided galactose. This improved GSL homeostasis in all patient-derived fibroblasts and in another SLC35A2 deficient cell model (CHO-Lec8). Additionally, SLC35A2-CDG serum analysis identified hydroxylated GSLs, particularly GM3, as potential disease biomarkers. Given the essential role of gangliosides in central nervous system function, their deficiency is likely a key factor in the neurological involvement of this disorder. These findings pave the way for new nutritional therapies with GSL supplements and highlight the importance of studying lipid glycosylation to better understand the complex pathophysiology of CDG.
Keywords: CDG; GSL; Gangliosides; Glycosphingolipids; SLC35A2-CDG; Tracer metabolomics.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval: The study was conducted in accordance with the Declaration of Helsinki and was approved by the ethical committee of the University Hospitals of Leuven. Fibroblast and serum analyses were performed in accordance with ethics application number S60206 (“Retrospective metabolic analysis of archived fibroblasts”) and S68513 (“Glycosphingolipid analysis in serum samples from individuals with inborn errors of metabolism”). Consent for publication: All the authors agree to publication. Competing interests: The authors have no relevant financial or non-financial interests to disclose.
Figures
References
-
- Kabuß R, Ashikov A, Oelmann S et al (2005) Endoplasmic reticulum retention of the large splice variant of the UDP-galactose transporter is caused by a Dilysine motif. Glycobiology 15:905–911. 10.1093/glycob/cwi085 - PubMed
-
- Dörre K, Olczak M, Wada Y et al (2015) A new case of UDP-galactose transporter deficiency (SLC35A2‐CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis 38:931–940. 10.1007/s10545-015-9828-6 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
