

Predicting Affinity Through Homology (PATH): Interpretable binding affinity prediction with persistent homology
- PMID: 40577377
- PMCID: PMC12226026
- DOI: 10.1371/journal.pcbi.1013216
Predicting Affinity Through Homology (PATH): Interpretable binding affinity prediction with persistent homology
Abstract
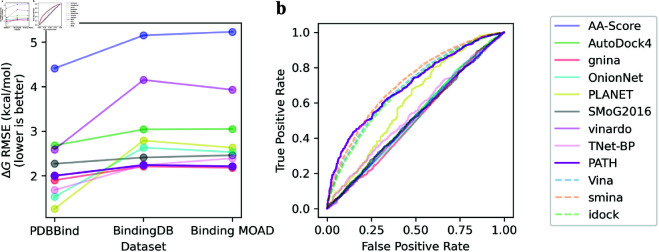
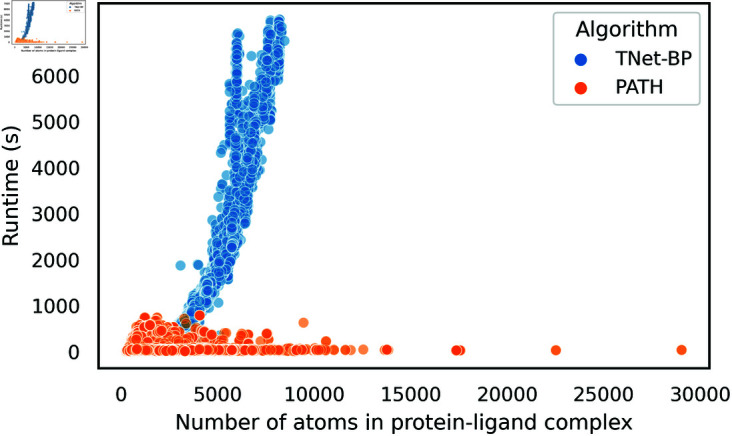
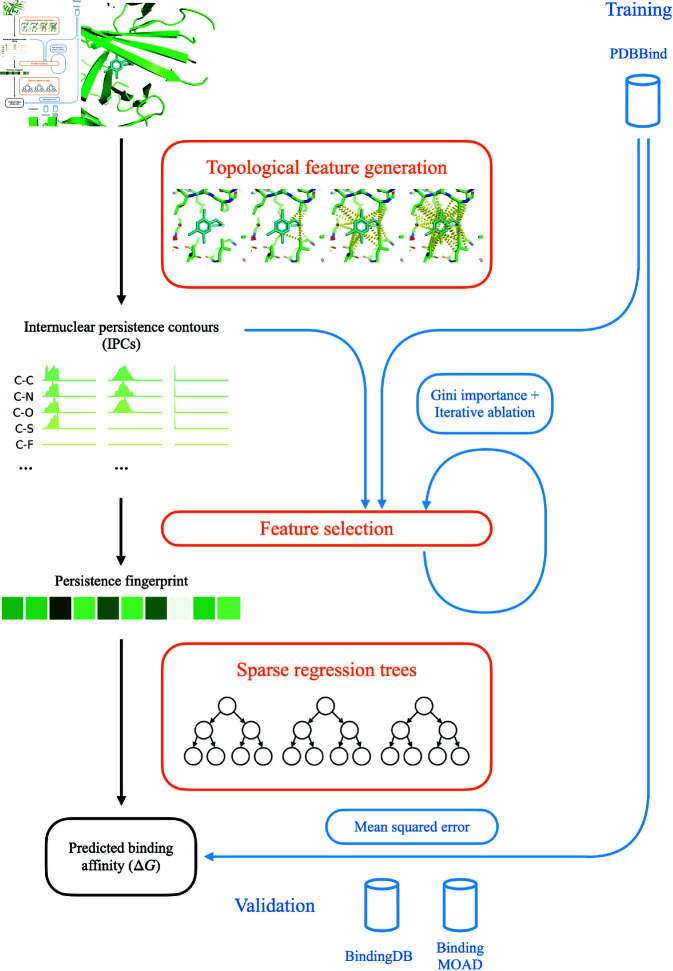
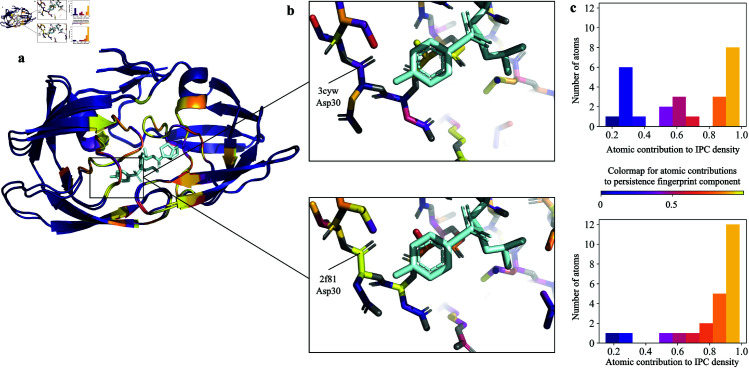
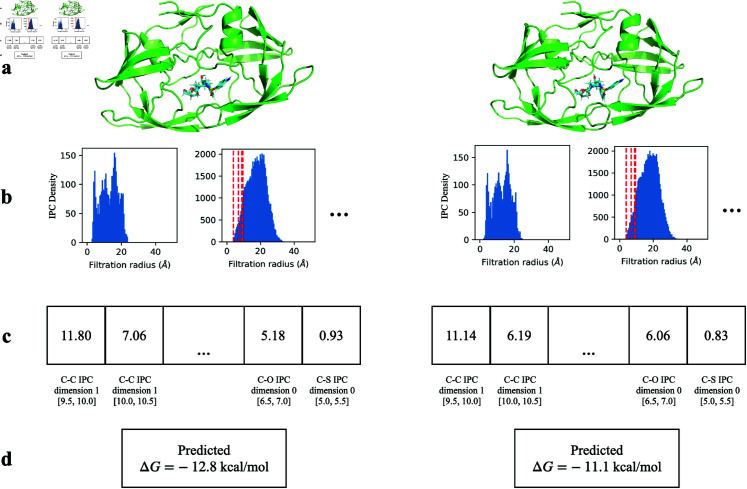
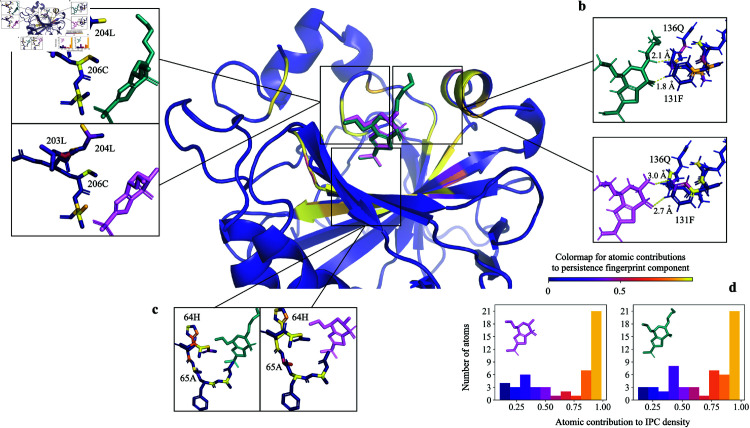
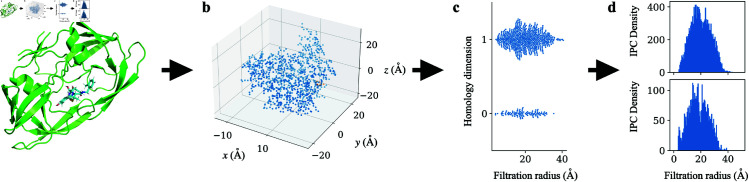
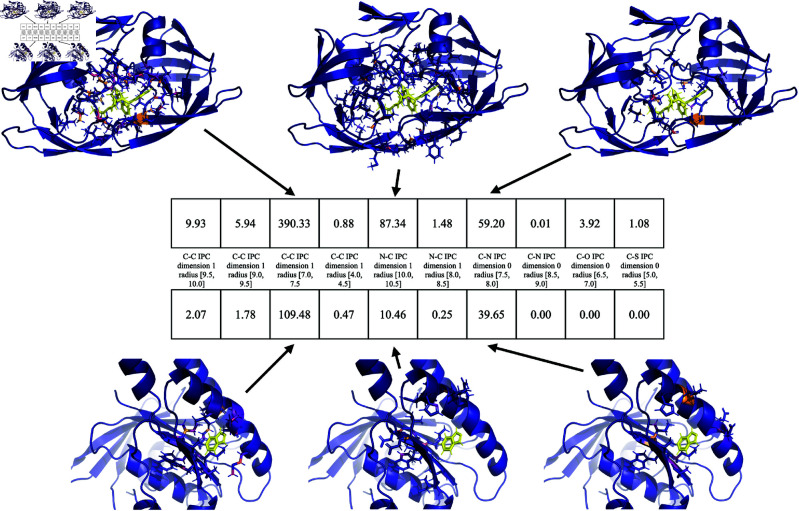
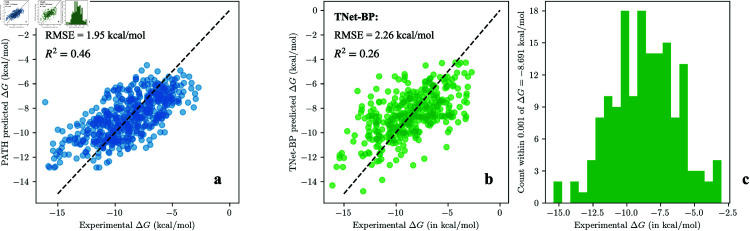
Accurate binding affinity prediction (BAP) is crucial to structure-based drug design. We present PATH+, a novel, generalizable machine learning algorithm for BAP that exploits recent advances in computational topology. Compared to current binding affinity prediction algorithms, PATH+ shows similar or better accuracy and is more generalizable across orthogonal datasets. PATH+ is not only one of the most accurate algorithms for BAP, it is also the first algorithm that is inherently interpretable. Interpretability is a key factor of trust for an algorithm and alongside generalizability, which allows PATH+ to be trusted in critical applications, such as inhibitor design. We visualized the features captured by PATH+ for two clinically relevant protein-ligand complexes and find that PATH+ captures binding-relevant structural mutations that are corroborated by biochemical data. Our work also sheds light on the features captured by current computational topology BAP algorithms that contributed to their high performance, which have been poorly understood. PATH+ also offers an improvement of 𝒪 (m + n)3 in computational complexity and is empirically over 10 times faster than the dominant (uninterpretable) computational topology algorithm for BAP. Based on insights from PATH+, we built PATH-, a scoring function for differentiating between binders and non-binders that has outstanding accuracy against 11 current algorithms for BAP. In summary, we report progress in a novel combination of interpretability, speed, and accuracy that should further empower topological screening of large virtual inhibitor libraries to protein targets, and allow binding affinity predictions to be understood and trusted. The source code for PATH+ and PATH- is released open-source as part of the OSPREY protein design software package.
Copyright: © 2025 Long, Donald. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: B.R.D. is a founder of Ten63 Therapeutics, Inc. B.R.D. was previously a guest editor for PLoS Comp. Biol.
Figures
Update of
-
Predicting Affinity Through Homology (PATH): Interpretable Binding Affinity Prediction with Persistent Homology.bioRxiv [Preprint]. 2024 Oct 21:2023.11.16.567384. doi: 10.1101/2023.11.16.567384. bioRxiv. 2024. Update in: PLoS Comput Biol. 2025 Jun 27;21(6):e1013216. doi: 10.1371/journal.pcbi.1013216. PMID: 38014181 Free PMC article. Updated. Preprint.
References
-
- Kontoyianni M. Docking and virtual screening in drug discovery. Proteomics for drug discovery: Methods and protocols. 2017. p. 255–66. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
