

Delineation of a thrombin receptor-stimulated vascular smooth muscle cell transition generating cells in the plaque-stabilizing fibrous cap
- PMID: 40577585
- PMCID: PMC12352313
- DOI: 10.1093/cvr/cvaf112
Delineation of a thrombin receptor-stimulated vascular smooth muscle cell transition generating cells in the plaque-stabilizing fibrous cap
Abstract
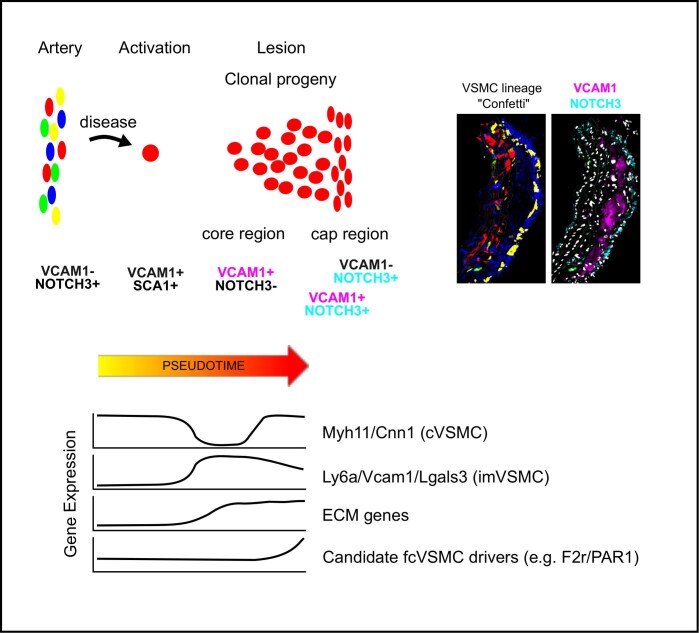
Aims: Vascular smooth muscle cells (VSMCs) accumulate in atherosclerotic plaques and exhibit remarkable phenotypic plasticity, contributing to both plaque growth and stability. The plaque-stabilizing fibrous cap is rich in VSMC-derived cells, yet the cellular transitions and regulatory mechanisms governing fibrous cap formation remain unclear. Here, we aimed to identify the VSMC phenotypic transitions associated with this critical process.
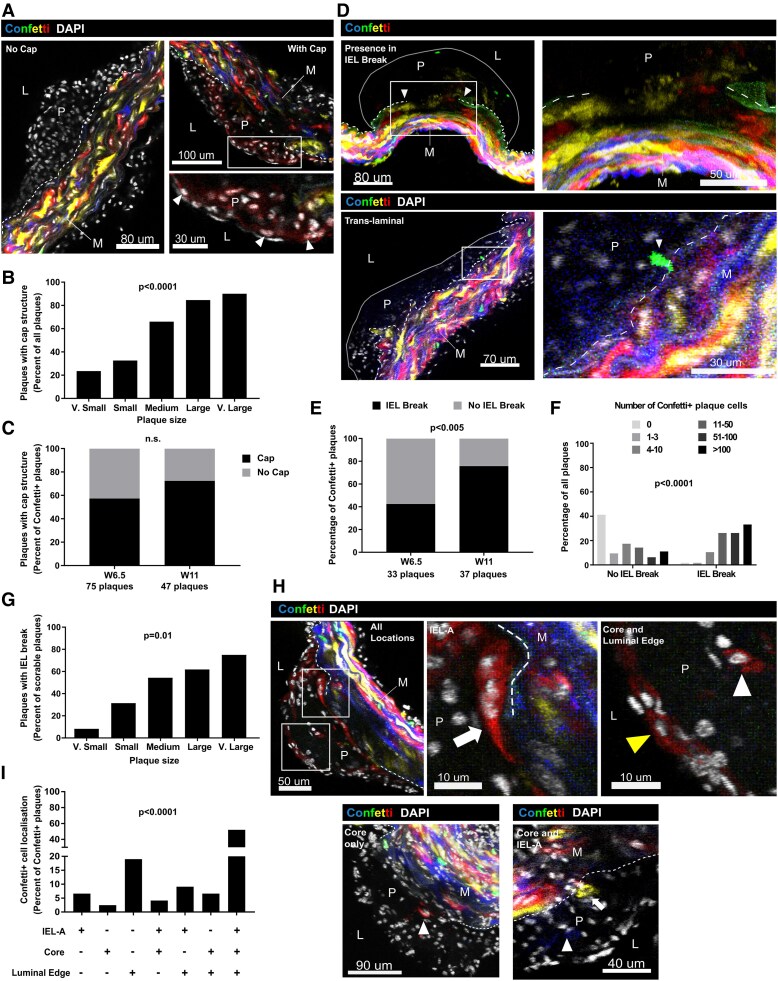
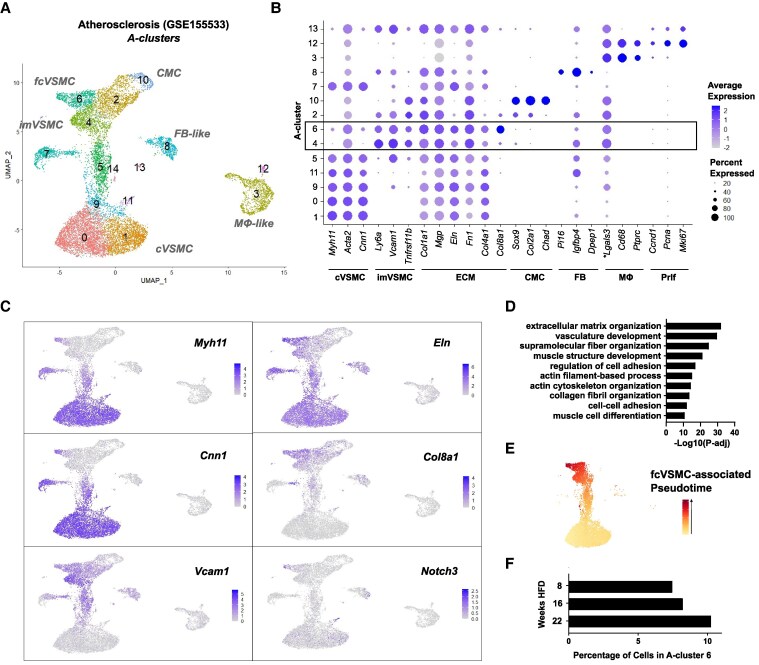
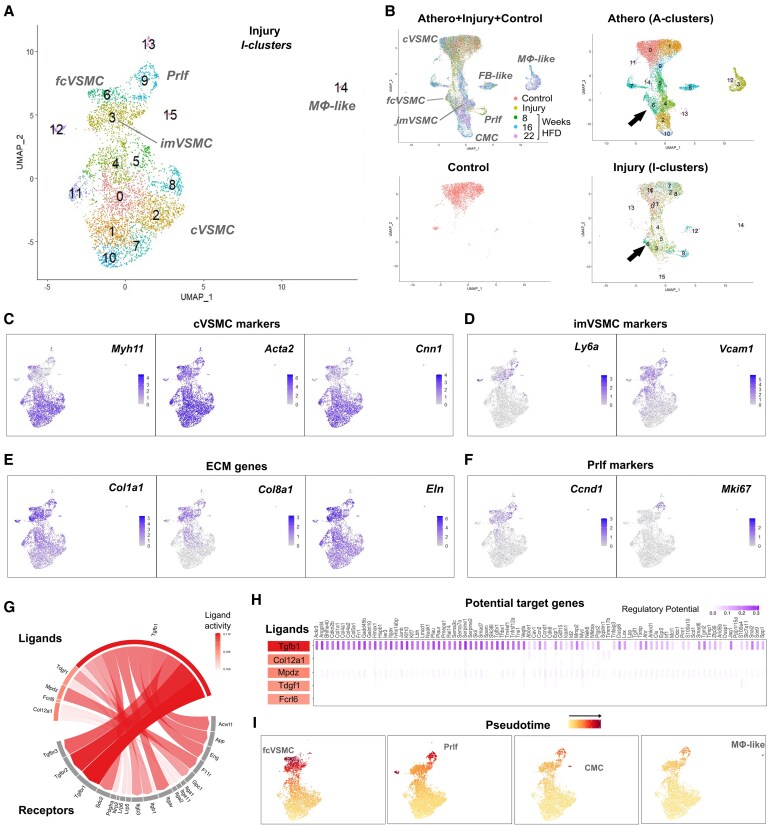
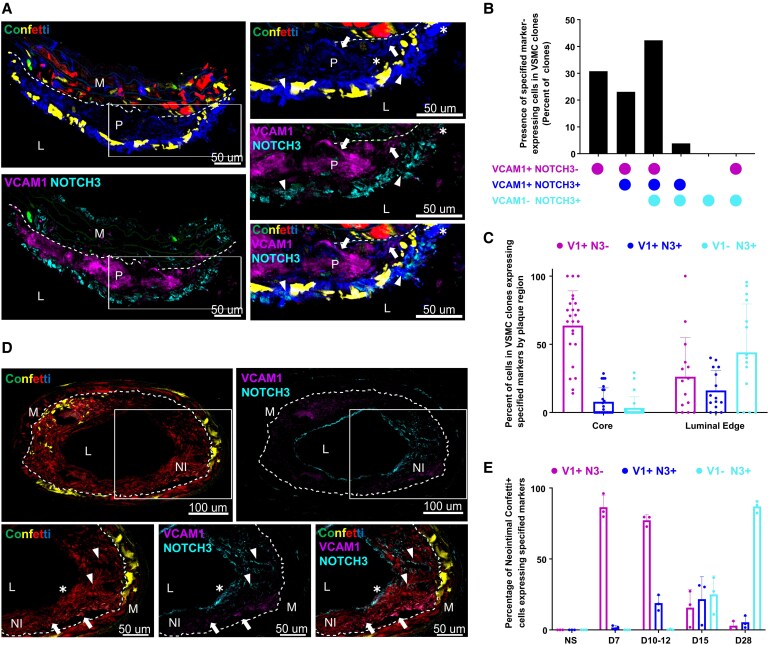
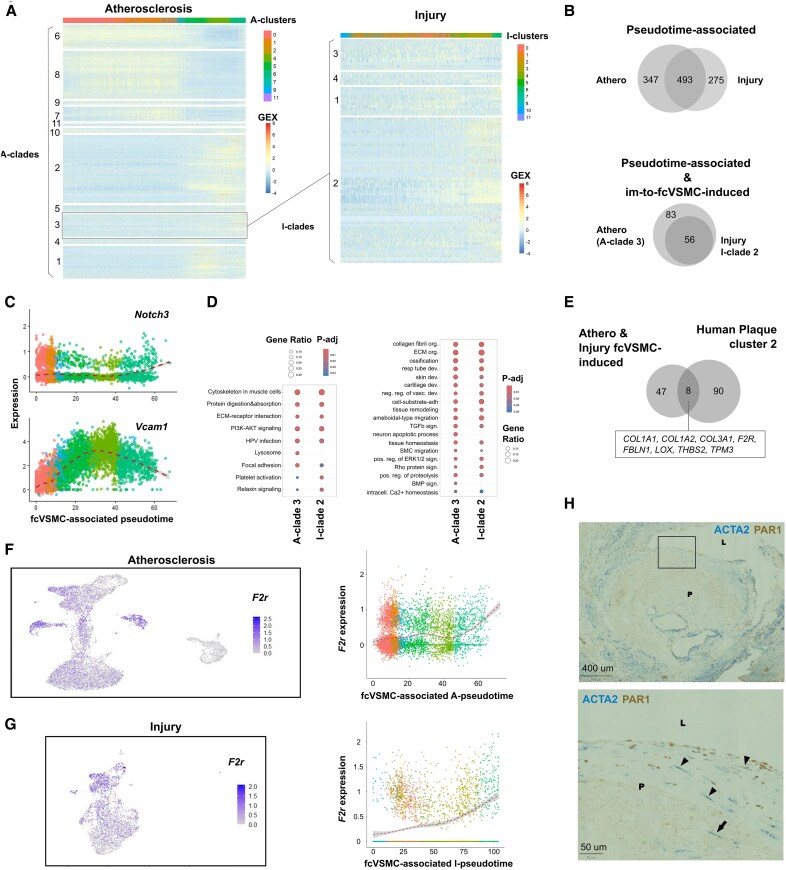
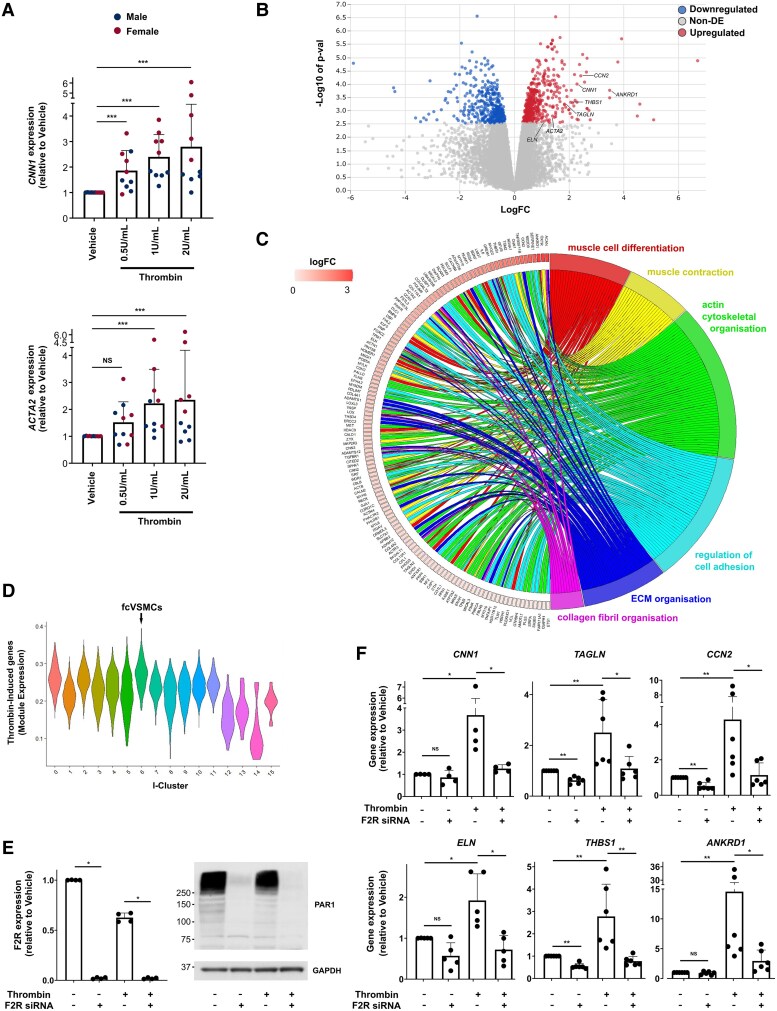
Methods and results: Mapping of lineage-traced VSMCs during plaque development revealed investment of VSMCs prior to fibrous cap formation. Using single-cell RNA-sequencing (scRNA-seq) profiles of lineage-traced VSMCs from atherosclerotic and acutely injured mouse arteries, we identified a disease-specific VSMC state co-expressing contractile genes with extracellular matrix (ECM) components (including fibrillar collagens and elastin) and NOTCH3, which are associated with fibrous cap formation. Computational trajectory analysis predicted that this proposed fibrous cap-related VSMC (fcVSMC) state arises from a previously described plastic, intermediate VSMC population expressing SCA1 and VCAM1. Clonal analysis further showed that NOTCH3+ fcVSMCs derive from intermediate VSMCs in both atherosclerosis and an acute vascular injury model, suggesting a conserved disease-relevant mechanism. The fcVSMCs were enriched in plaque fibrous caps compared to lesion cores, consistent with a role in fibrous cap formation. By combining scRNA-seq trajectory analysis and spatial transcriptomics of human atherosclerotic plaques, we identified protease-activated receptor-1 (PAR1) as a candidate regulator of fcVSMC generation. PAR1 was expressed by VSMCs in human plaque fibrous caps and PAR1 activation by thrombin induced expression of contractile genes and ECM components associated with the fcVSMC state in human VSMCs.
Conclusion: Our findings identify a VSMC transition linked to fibrous cap formation in atherosclerosis and show this is modelled by vascular injury. We identify VSMC-expressed PAR1 as a potential therapeutic target for promoting plaque stability by driving the transition to the matrix-producing, fibrous cap-associated VSMC state.
Keywords: Atherosclerosis; Fibrous cap; Phenotypic switching; Protease-activated receptor-1; Vascular smooth muscle cells.
© The Author(s) 2025. Published by Oxford University Press on behalf of the European Society of Cardiology.
Conflict of interest statement
Conflict of interest: H.F.J. is a key opinion leader for and is funded by Novo Nordisk A/S.
Figures
Similar articles
-
Trimethylamine-N-oxide affects cell type-specific pathways and networks in mouse aorta to promote atherosclerotic plaque vulnerability.bioRxiv [Preprint]. 2025 Mar 1:2025.02.25.640205. doi: 10.1101/2025.02.25.640205. bioRxiv. 2025. Update in: Arterioscler Thromb Vasc Biol. 2025 Aug 14. doi: 10.1161/ATVBAHA.125.323047. PMID: 40060573 Free PMC article. Updated. Preprint.
-
Vascular Smooth Muscle Cells: A Therapeutic Target in Atherosclerosis.Rev Cardiovasc Med. 2025 Jun 26;26(6):28240. doi: 10.31083/RCM28240. eCollection 2025 Jun. Rev Cardiovasc Med. 2025. PMID: 40630429 Free PMC article. Review.
-
Insulin receptors in vascular smooth muscle cells regulate plaque stability of atherosclerosis.Cardiovasc Res. 2024 Dec 14;120(16):2017-2030. doi: 10.1093/cvr/cvae193. Cardiovasc Res. 2024. PMID: 39197028
-
Trimethylamine-N-Oxide Affects Cell Type-Specific Pathways and Networks in Mouse Aorta to Promote Atherosclerotic Plaque Vulnerability.Arterioscler Thromb Vasc Biol. 2025 Aug 14. doi: 10.1161/ATVBAHA.125.323047. Online ahead of print. Arterioscler Thromb Vasc Biol. 2025. PMID: 40808653
-
IL-1β inhibition in stabilizing atherosclerotic plaques: The critical role of fibroblast-like cells.Vascul Pharmacol. 2025 Jun;159:107493. doi: 10.1016/j.vph.2025.107493. Epub 2025 Apr 1. Vascul Pharmacol. 2025. PMID: 40180256 Review.
References
-
- Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 2019;16:727–744. - PubMed
-
- Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S, Weiser-Evans MCM. SDF-1a; induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol 2011;31:1300–1308. - PMC - PubMed
Grants and funding
- NIHR BRC
- 2018-190766/RG98793/Chan Zuckerberg Initiative
- CH/2000003/12800/BHF_/British Heart Foundation/United Kingdom
- RM/13/3/30159/BHF_/British Heart Foundation/United Kingdom
- National Institute of Heath Research Cambridge Biomedical Research Centre
- WT_/Wellcome Trust/United Kingdom
- RE/13/6/30180/BHF_/British Heart Foundation/United Kingdom
- FS/15/38/31516/BHF_/British Heart Foundation/United Kingdom
- Collaborative Bioresource for Translational Medicine
- PG/19/6/34153/BHF_/British Heart Foundation/United Kingdom
- RE/18/1/34212/BHF_/British Heart Foundation/United Kingdom
- FS/15/62/32032/BHF_/British Heart Foundation/United Kingdom
LinkOut - more resources
Full Text Sources
Miscellaneous
