

Genetic transcriptional regulation profiling of cartilage reveals pathogenesis of osteoarthritis
- PMID: 40577937
- PMCID: PMC12278629
- DOI: 10.1016/j.ebiom.2025.105821
Genetic transcriptional regulation profiling of cartilage reveals pathogenesis of osteoarthritis
Abstract
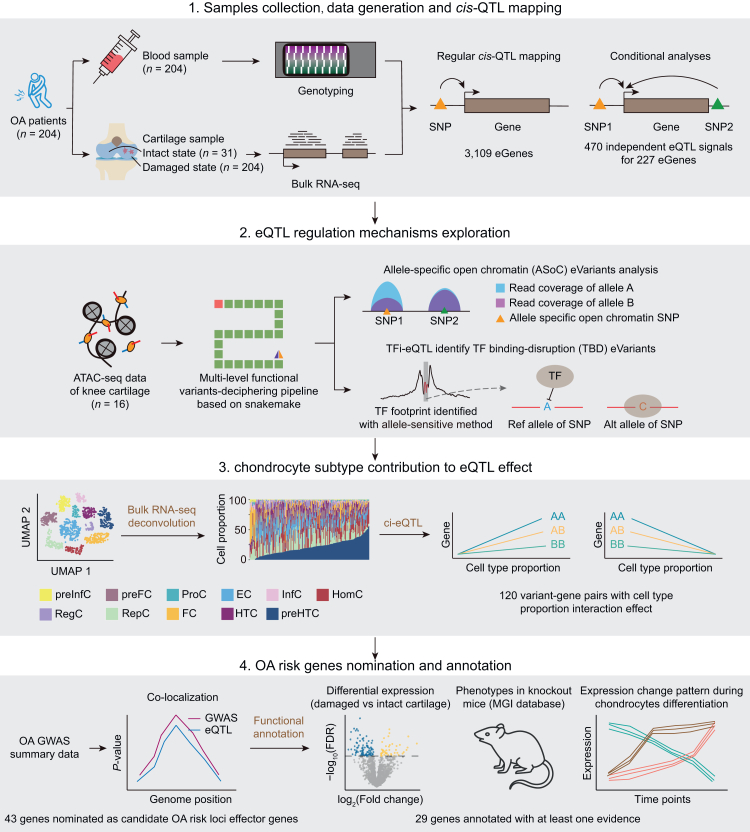
Background: Genome-wide association studies (GWAS) have identified more than one hundred risk loci for osteoarthritis (OA). Identifying the effector genes and deciphering the underlying regulatory mechanisms are of great importance but remains challenging due to limited availability of OA-related tissue data. This study aims to address this issue by generating a cartilage expression quantitative trait loci (eQTLs) and a functional fine-mapping resource.
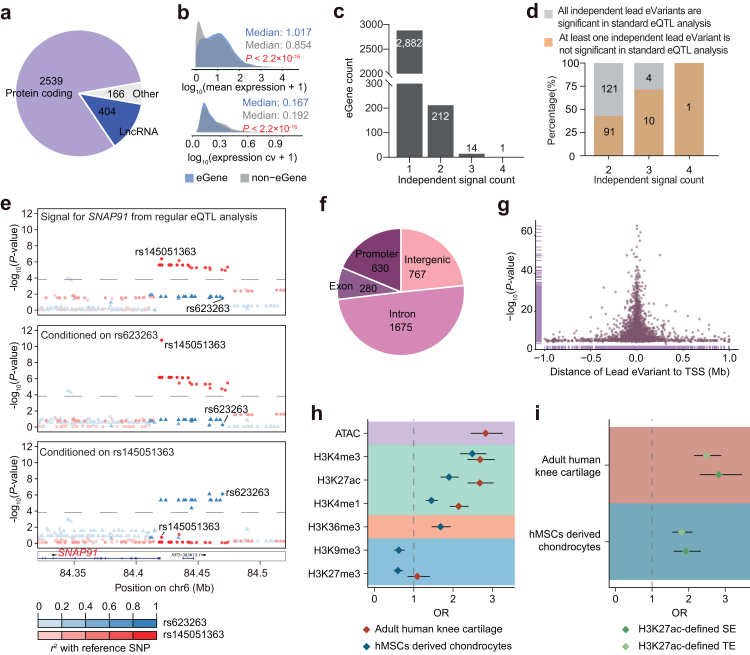
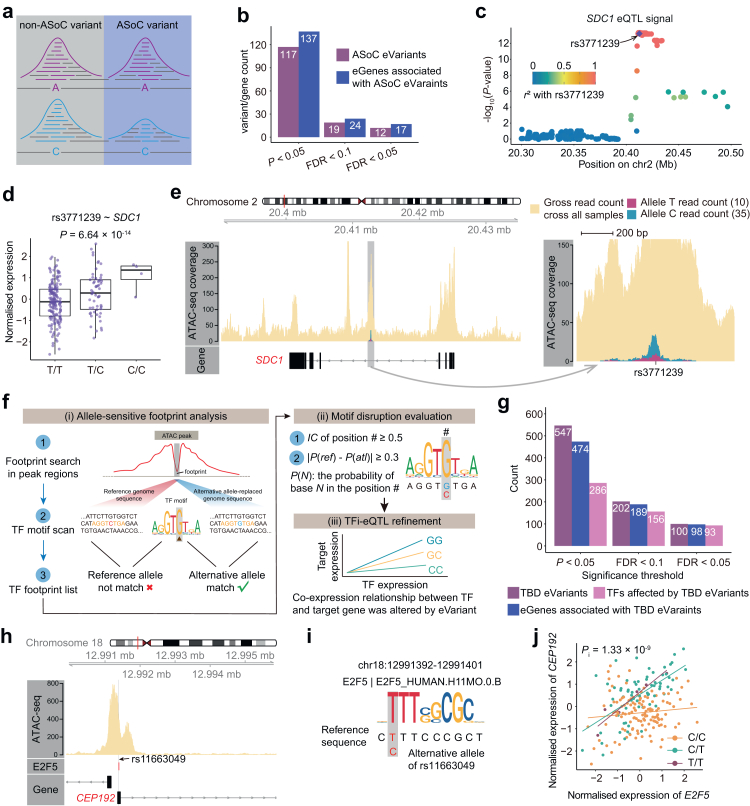
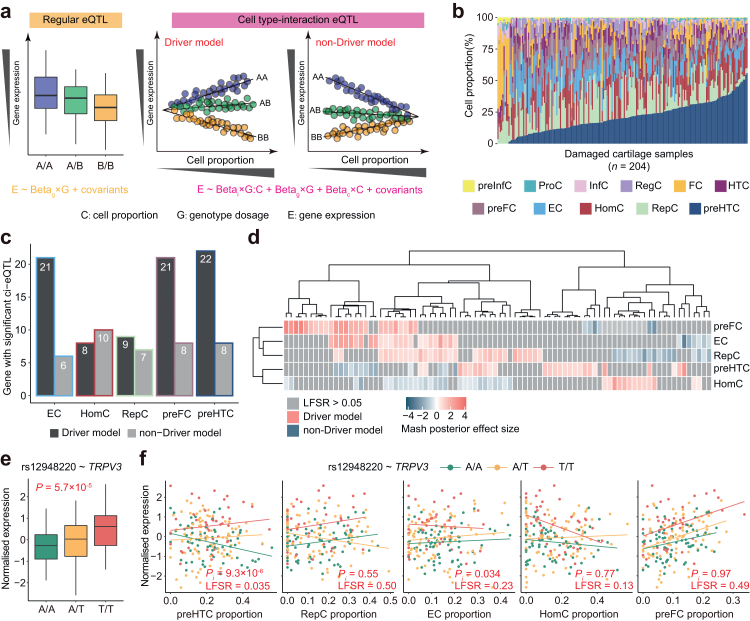
Methods: We performed cis-eQTL analysis using genomics and cartilage transcriptomics data from 204 patients with OA (largest sample size to date). Cell type-interaction eQTL analysis (ci-eQTL) was conducted to explore the chondrocyte subtype dependency of eQTL effects. Co-localization analysis was used to nominate effector genes of OA GWAS risk loci. A deciphering pipeline was established to identify candidate causal variants in eQTL loci that regulate gene expression through the alteration of chromatin accessibility or disruption of transcription factors (TFs) binding to regulatory elements.
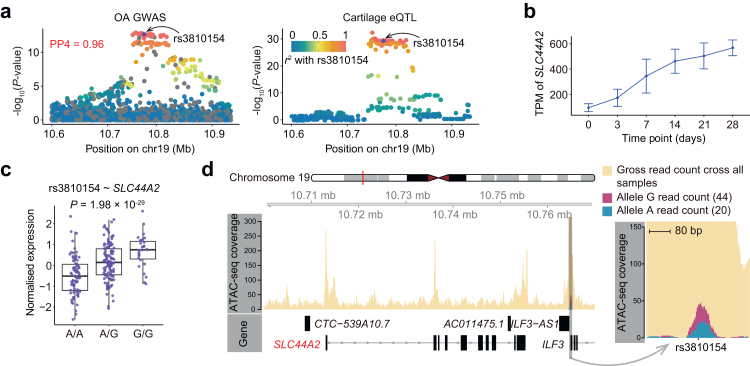
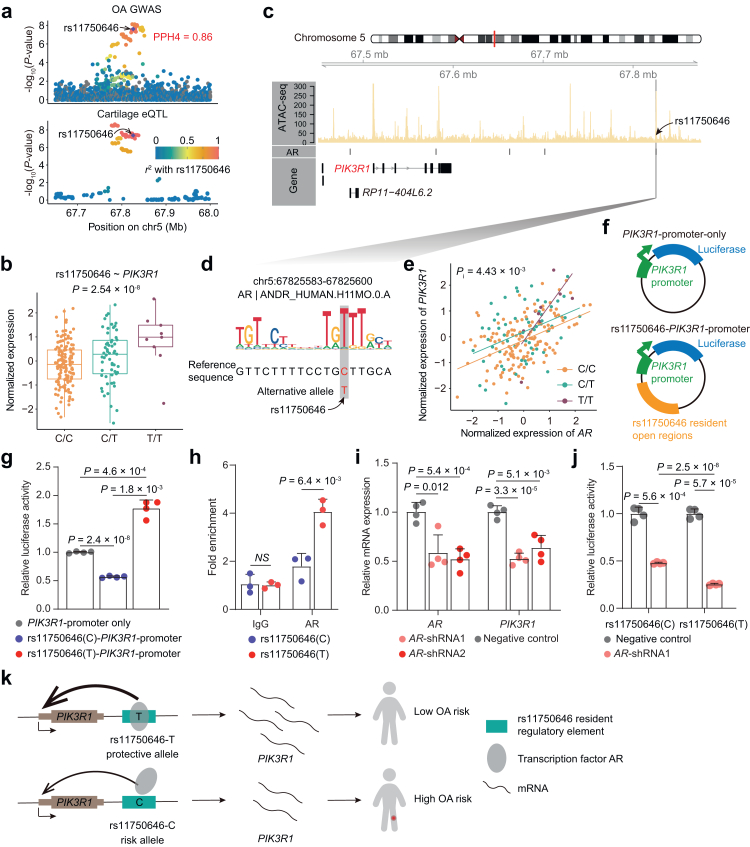
Findings: We identified 3352 independent eQTLs for 3109 genes, 120 eQTL-gene pairs showed chondrocyte subtype dependency. We identified 19 new OA risk genes. We identified 117 causal eQTLs exhibiting allele-specific open chromatin (ASoC) and 547 eQTLs involved in transcription factor binding disruption (TBD). Functional validation showed that the T allele of the OA risk variant rs11750646 enhances the AR binding affinity to an open chromatin region, thereby promoting the expression of the OA-related gene PIK3R1.
Interpretation: Our findings provide insights into the unique regulatory landscape of cartilage and elucidate potential mechanisms underlying OA pathogenesis.
Funding: This work was supported by National Natural Science Foundation of China (32470639, 82372458, and 82170896); Science Fund for Distinguished Young Scholars of Shaanxi Province (2025JC-JCQN-054); Innovation Capability Support Program of Shaanxi Province (2022TD-44, 2024RS-CXTD-86); Key Research and Development Project of Shaanxi Province (2023-YBSF-180); China Postdoctoral Science Foundation (2024M752561); and the Fundamental Research Funds for the Central Universities.
Keywords: Cartilage; Fine-mapping; Osteoarthritis; eQTL.
Copyright © 2025 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Hunter D.J., Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–1759. - PubMed
-
- Warner S.C., Valdes A.M. The genetics of osteoarthritis: a review. J Funct Morphol Kinesiol. 2016;1(1):140–153.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
