

A bedside-to-bench translational analysis of NF1 alterations and CDK4/6 inhibitor resistance in hormone receptor-positive metastatic breast cancer
- PMID: 40578027
- PMCID: PMC12266523
- DOI: 10.1016/j.ebiom.2025.105828
A bedside-to-bench translational analysis of NF1 alterations and CDK4/6 inhibitor resistance in hormone receptor-positive metastatic breast cancer
Abstract
Background: CDK4/6 inhibitors (CDK4/6i) are used for management of hormone receptor-positive (HR+) metastatic breast cancer (MBC), and activation of the RAS/MAPK and PI3K/AKT signalling pathways has been implicated in resistance to these agents. Pathogenic NF1 mutations (pNF1m) dysregulate RAS signalling, but NF1 has not been linked to CDK4/6i resistance. We analysed multi-institutional data, real-world evidence, and preclinical models to characterise the impact of pNF1m on CDK4/6i sensitivity.
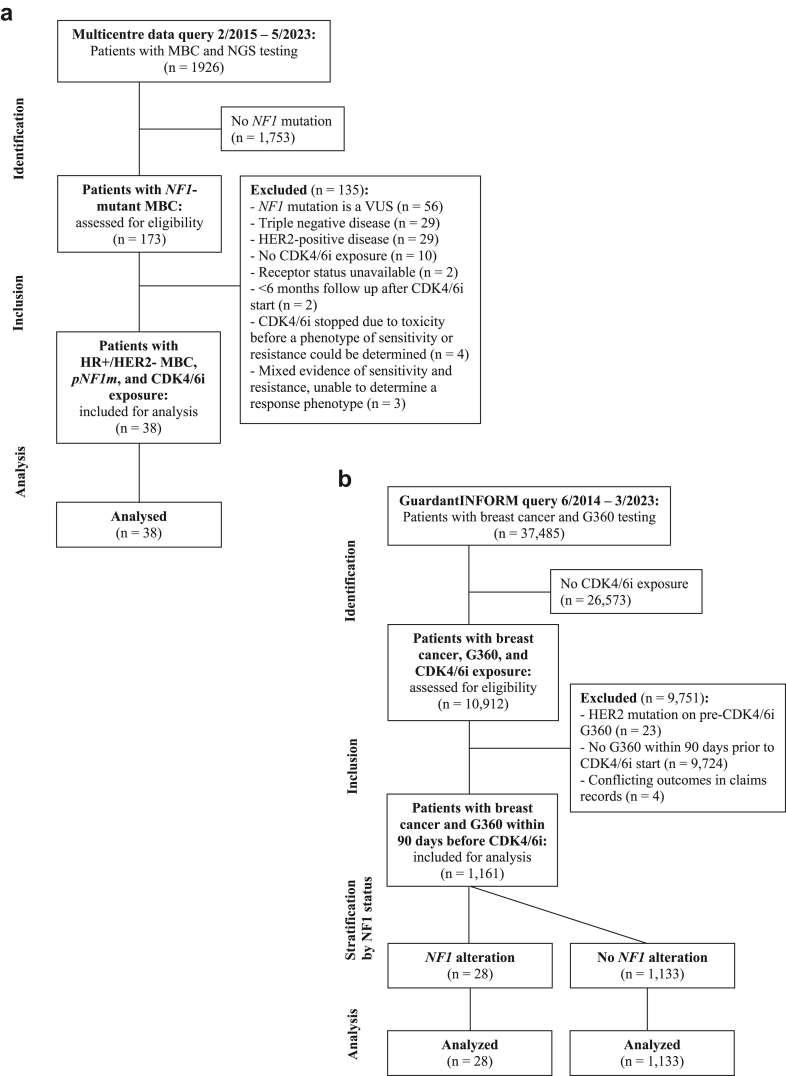
Methods: A retrospective cohort of patients with pNF1m tumours were identified from 4 institutions between 2/2015-5/2023 and evaluated for progression-free survival and intrinsic/acquired resistance on CDK4/6i. Real-world clinical-genomic data from GuardantINFORM between 6/2014 and 3/2023 was analysed for associations between pNF1m and time-to-next-treatment or overall survival following CDK4/6i, adjusted using propensity score weighting. We used CRISPR/Cas9 to delete NF1 in MCF7 and T47D breast cancer cells in vitro. NF1-knockout (NF1-KO) and -wild-type (WT) cells were analysed with respect to CDK4/6i sensitivity, MAPK and PI3K pathway activation, and sensitivity to MAPK and PI3K pathway inhibitors. In parallel, we assessed treatment response in a patient-derived organoid (PDO) harbouring NF1 loss, established from an HR+/HER2- breast tumor following progression on a CDK4/6i.
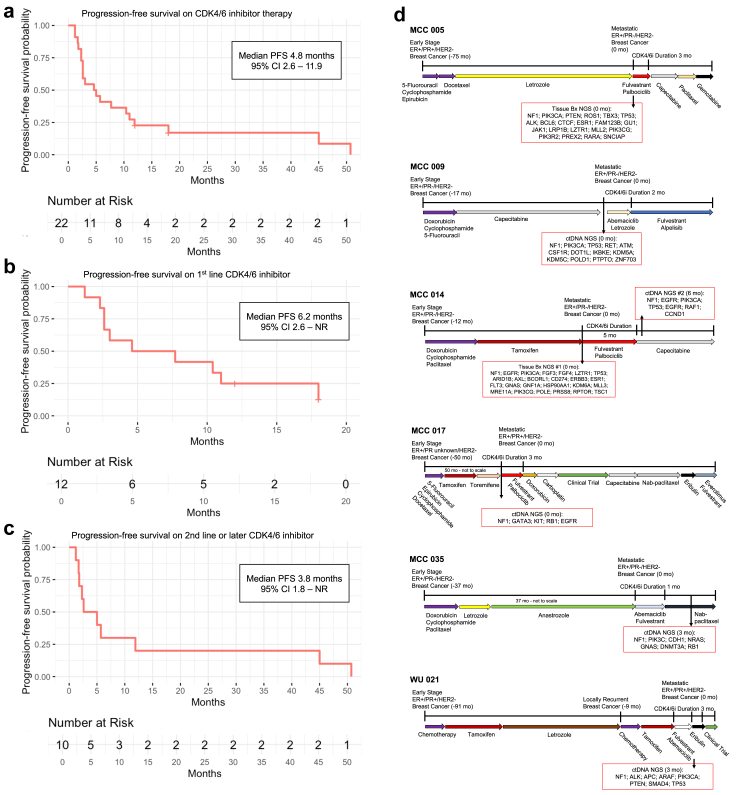
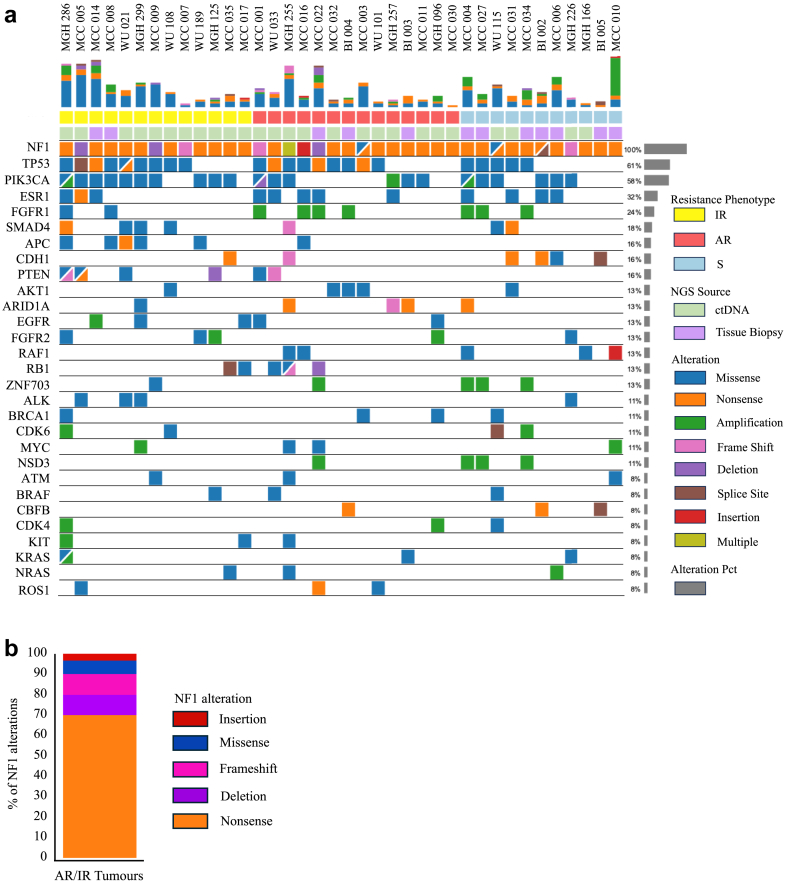
Findings: Among 1962 multicentre patients, we identified 38 with HR+/HER2- MBC, pNF1m, and exposure to CDK4/6i. NF1-associated intrinsic or acquired resistance to CDK4/6i was observed in a majority of tumours, and in those with baseline pNF1m on first-line CDK4/6i, a median progression-free survival of 6.2 months was much less than expected in routine practice. Real-world weighted analysis of 1161 patients comparing 28 pNF1m to 1133 NF1 non-altered tumours demonstrated shorter time-to-next-treatment on CDK4/6i regimens (4.2 vs. 12.4 months, hazard ratio 3.14, 95% confidence interval 2.01-4.93) and overall survival (15.8 vs. 45.2 months, hazard ratio 2.04, 95% confidence interval 1.09-3.82). NF1-deleted cells exhibited reduced sensitivity to CDK4/6i with or without oestrogen suppression, which was accompanied by induction of both MAPK and PI3K pathways, the latter of which was exacerbated by CDK4/6i. Blockade of RAS or AKT, but not MEK or ERK, reversed CDK4/6i resistance mediated by NF1 loss in cell lines and the PDO.
Interpretation: NF1 mutations are associated with shorter therapy duration on CDK4/6i in MBC. A causal link between NF1 loss and CDK4/6i resistance was supported by experiments in HR + breast cancer cells. NF1 deletion was accompanied by activation of ERK and AKT, and blockade of RAS or AKT combined with CDK4/6i was effective in NF1-deleted cells and an NF1-mutant PDO.
Funding: Breast Cancer Research Foundation DRC-20-001, National Cancer Institute R01CA273246, National Institute of Health P30 CA142543, Susan G. Komen Breast Cancer Foundation SAB1800010, Department of Defence BC 210406, Mary Kay Ash Foundation International Postdoctoral Scholars in Cancer Research Fellowship.
Keywords: CDK4/6 inhibitor; HR+ breast cancer; Metastatic breast cancer; NF1 mutation; Precision medicine; Resistance mechanisms.
Copyright © 2025 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests Maxwell R. Lloyd: None. Rosario Chica-Parrado: None. Caroline M. Weipert: Employee/stockholder: Guardant Health, Inc. Todd C. Knepper: Consulting/Advisory Board: AstraZeneca. Emily L. Podany: None. Fabiana Napolitano: None. Dan Ye: None. Chang-Ching Lin: None. Yasuaki Uemoto: None. Jiemin Liao: Employee/stockholder: Guardant Health, Inc. Claire Wegrzyn: None. Christine M. Walko: None. Lianne Y. Ryan: None. Jennifer C. Keenan: None. Arielle J. Medford: Consulting/Advisory Board: AstraZeneca, Guardant Health, Illumina, Myriad Genetics, Science for America Speaking/Education: Natera. Shiyuan A. Liu: None. Gerburg M. Wulf: Research funding from Mersana, Gilead, Seagen, Celcuity, Totus Medicines, Agios, Nikang (institutional funding unrelated to this project), and US patent 20090258352, “A1 Pin 1 as a marker for abnormal cell growth,” licenced to Cell Signalling and R&D Systems. Katherine K. Clifton: None. Cynthia X. Ma: Honoraria: PlusOne Health GmbH, Guardant Health. Consulting or Advisory Role: Novartis, Seagen, Agendia, AstraZeneca, Athenex, Bayer HealthCare Pharmaceuticals, Biovica Inc, Olaris, Sanofi- Genzyme, Gilead Sciences, Pfizer, Lilly, Tempus. Research Funding: Pfizer (Inst), Puma Biotechnology (Inst). Hyo S. Han: Research funding to institution from AbbVie, Arvinas, GSK, G1 Therapeutics, Quantum Leap Healthcare Collaborative, Marker, Pfizer, Zymeworks, Celcuity, and Department of Defence; payment for speakers bureaus from Lilly; advisory board for Novartis, AstraZeneca, and Gilead. Nicole Zhang: Employee/stockholder: Guardant Health, Inc. Leif W. Ellisen: Consultant: Kisoji Biotechnology and Mersana Therapeutics. Aditya Bardia: Consultant/Advisory board: Pfizer, Novartis, Genentech, Merck, Radius Health, Immunomedics/Gilead, Sanofi, Daiichi Pharma/Astra Zeneca, Phillips, Eli Lilly, Foundation Medicine. Contracted Research/Grant (to institution): Genentech, Novartis, Pfizer, Merck, Sanofi, Radius Health, Immunomedics/Gilead, Daiichi Pharma/Astra Zeneca, Eli Lilly. Carlos L. Arteaga: CLA has served as scientific advisor to Novartis, Lilly, Merck, Daiichi Sankyo, AstraZeneca, Sanofi, OrigiMed, PUMA Biotechnology, Immunomedics, Athenex, Arvinas, and the Susan G. Komen Foundation; has received grant support from Pfizer, Lilly, and Takeda; and holds minor stock options in Provista. Ariella B. Hanker: Consulting: Trishula; Research funding: Breast Cancer Research Foundation/Lilly drug research collaborative. Seth A. Wander: Consulting/Advisory Board: Foundation Medicine, Veracyte, Eli Lilly, Pfizer, Novartis, AstraZeneca, Hologic, Biovica, Puma Biotechnology, Regor Pharmaceuticals; Speaking/Education: Eli Lilly, 2ndMD, Guardant Health; Institutional Research Support: Genentech, Eli Lilly, Pfizer, Nuvation Bio, Regor Pharmaceuticals.
Figures



References
-
- Cichowski K., Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. - PubMed
-
- Pearson A., Proszek P., Pascual J., et al. Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin Cancer Res. 2020;26:608–622. - PubMed
-
- Lau N., Feldkamp M.M., Roncari L., et al. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol. 2000;59:759–767. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
