

GRAPEVNE - Graphical Analytical Pipeline Development Environment for Infectious Diseases
- PMID: 40585005
- PMCID: PMC12205304
- DOI: 10.12688/wellcomeopenres.23824.1
GRAPEVNE - Graphical Analytical Pipeline Development Environment for Infectious Diseases
Abstract
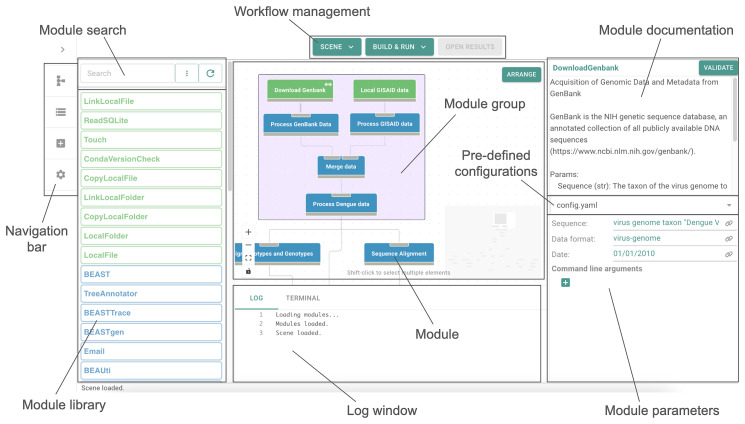
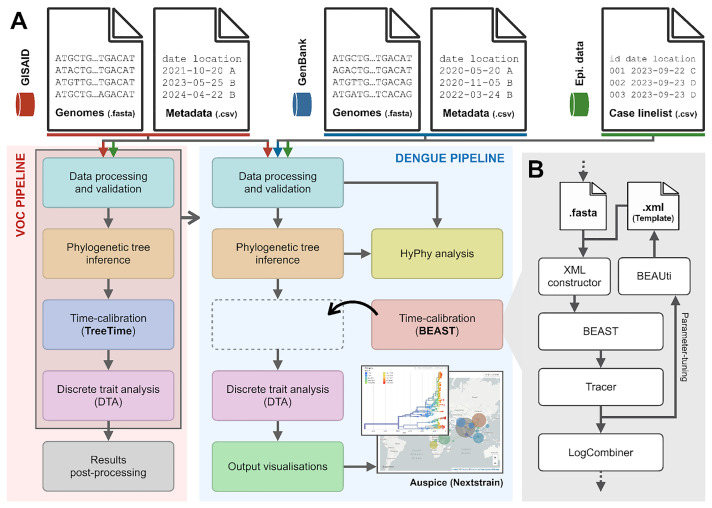
The increase in volume and diversity of relevant data on infectious diseases and their drivers provides opportunities to generate new scientific insights that can support 'real-time' decision-making in public health across outbreak contexts and enhance pandemic preparedness. However, utilising the wide array of clinical, genomic, epidemiological, and spatial data collected globally is difficult due to differences in data preprocessing, data science capacity, and access to hardware and cloud resources. To facilitate large-scale and routine analyses of infectious disease data at the local level (i.e. without sharing data across borders), we developed GRAPEVNE (Graphical Analytical Pipeline Development Environment), a platform enabling the construction of modular pipelines designed for complex and repetitive data analysis workflows through an intuitive graphical interface. Built on the Snakemake workflow management system, GRAPEVNE streamlines the creation, execution, and sharing of analytical pipelines. Its modular approach already supports a diverse range of scientific applications, including genomic analysis, epidemiological modeling, and large-scale data processing. Each module in GRAPEVNE is a self-contained Snakemake workflow, complete with configurations, scripts, and metadata, enabling interoperability. The platform's open-source nature ensures ongoing community-driven development and scalability. GRAPEVNE empowers researchers and public health institutions by simplifying complex analytical workflows, fostering data-driven discovery, and enhancing reproducibility in computational research. Its user-driven ecosystem encourages continuous innovation in biomedical and epidemiological research but is applicable beyond that. Key use-cases include automated phylogenetic analysis of viral sequences, real-time outbreak monitoring, forecasting, and epidemiological data processing. For instance, our dengue virus pipeline demonstrates end-to-end automation from sequence retrieval to phylogeographic inference, leveraging established bioinformatics tools which can be deployed to any geographical context. For more details, see documentation at: https://grapevne.readthedocs.io.
Keywords: automated workflows; data science; epidemiology; genomics; graphical interface; open-source; outbreaks; snakemake.
Plain language summary
With the growing amount of data on infectious diseases, researchers have new opportunities to improve public health decisions and pandemic preparedness. However, analyzing this vast and diverse data—spanning clinical records, genomic sequences, epidemiological trends, and geographic information—can be challenging due to differences in data processing methods, technical expertise, and access to computing resources. To address these challenges, we developed GRAPEVNE, a user-friendly platform that helps researchers build and manage complex data analysis workflows using a visual interface. Built on the Snakemake workflow management system, GRAPEVNE simplifies the process of organizing and running large-scale studies, making it easier to track outbreaks, analyze disease patterns, and process health data efficiently. Its modular approach allows users to customize workflows based on their specific needs, ensuring flexibility and ease of use. As an open-source platform, GRAPEVNE fosters collaboration and rolling development, supporting a wide range of applications, including genomic analysis, epidemiological modeling, and outbreak monitoring. Researchers can use it for tasks such as studying viral evolution, predicting disease spread, and processing epidemiological data across different geographical contexts. By streamlining data analysis, GRAPEVNE empowers public health institutions and researchers to make data-driven decisions more effectively. For more details, visit: https://grapevne.readthedocs.io.
Copyright: © 2025 Brittain JS et al.
Conflict of interest statement
No competing interests were disclosed.
Figures
Similar articles
-
How lived experiences of illness trajectories, burdens of treatment, and social inequalities shape service user and caregiver participation in health and social care: a theory-informed qualitative evidence synthesis.Health Soc Care Deliv Res. 2025 Jun;13(24):1-120. doi: 10.3310/HGTQ8159. Health Soc Care Deliv Res. 2025. PMID: 40548558
-
Heron, a Knowledge Graph editor for intuitive implementation of Python-based experimental pipelines.Elife. 2025 Jul 16;13:RP91915. doi: 10.7554/eLife.91915. Elife. 2025. PMID: 40667891 Free PMC article.
-
Accreditation through the eyes of nurse managers: an infinite staircase or a phenomenon that evaporates like water.J Health Organ Manag. 2025 Jun 30. doi: 10.1108/JHOM-01-2025-0029. Online ahead of print. J Health Organ Manag. 2025. PMID: 40574247
-
How to Implement Digital Clinical Consultations in UK Maternity Care: the ARM@DA Realist Review.Health Soc Care Deliv Res. 2025 May;13(22):1-77. doi: 10.3310/WQFV7425. Health Soc Care Deliv Res. 2025. PMID: 40417997 Review.
-
The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher's disease: a systematic review.Health Technol Assess. 2006 Jul;10(24):iii-iv, ix-136. doi: 10.3310/hta10240. Health Technol Assess. 2006. PMID: 16796930
References
-
- UK Health Security Agency: COVID-19 variants identified in the UK – latest updates. GOV.UK,2021. Reference Source
LinkOut - more resources
Full Text Sources
Research Materials
