

This is a preprint.
Boosting AlphaFold Protein Tertiary Structure Prediction through MSA Engineering and Extensive Model Sampling and Ranking in CASP16
- PMID: 40585263
- PMCID: PMC12204356
- DOI: 10.21203/rs.3.rs-6845168/v1
Boosting AlphaFold Protein Tertiary Structure Prediction through MSA Engineering and Extensive Model Sampling and Ranking in CASP16
Abstract
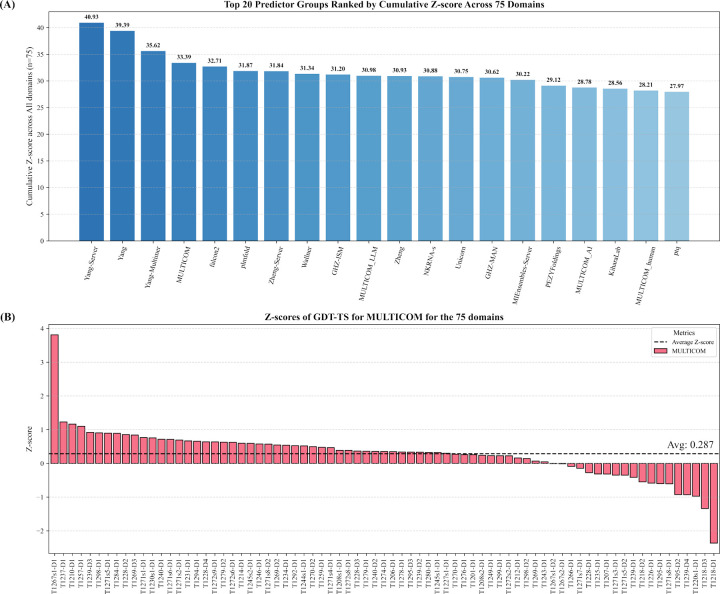
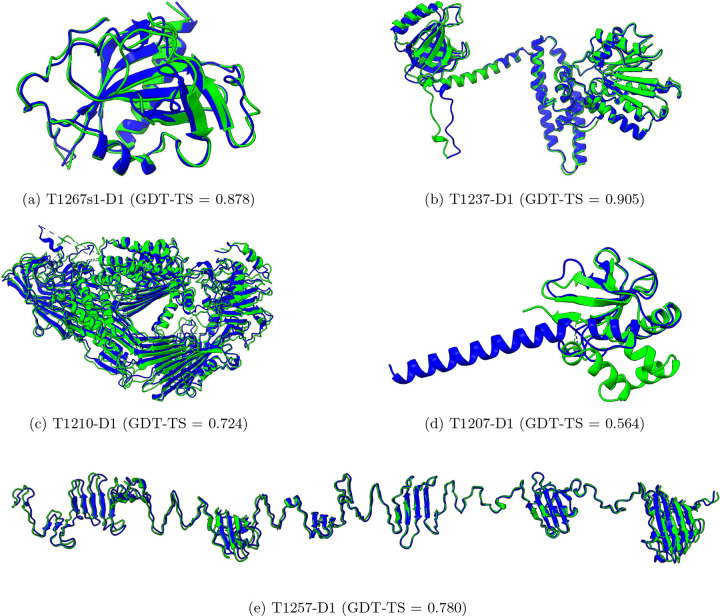
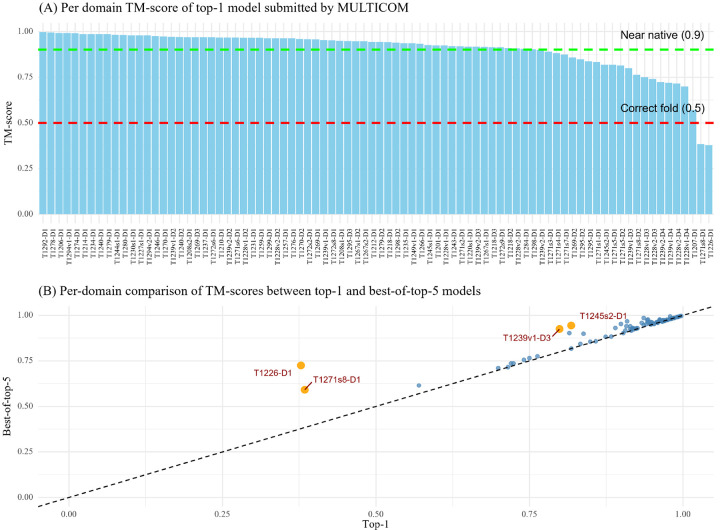
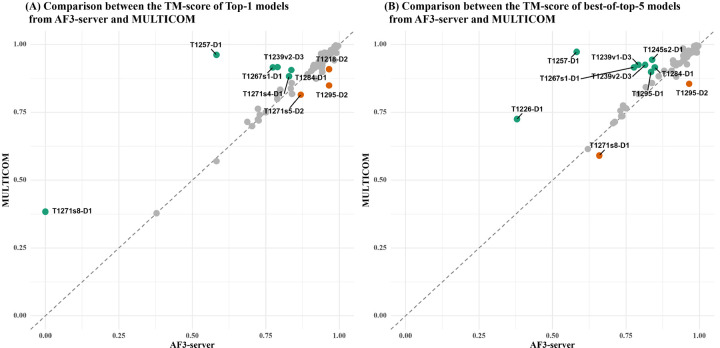
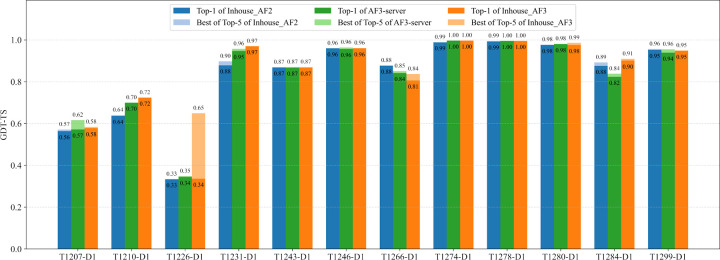
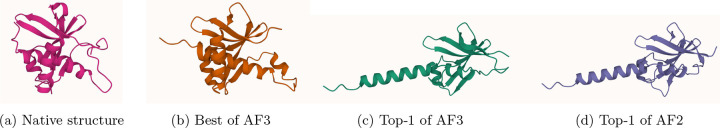
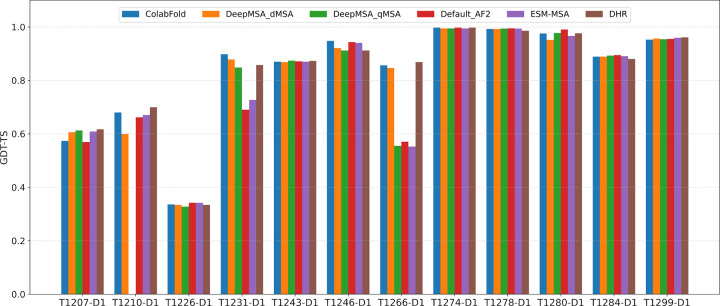
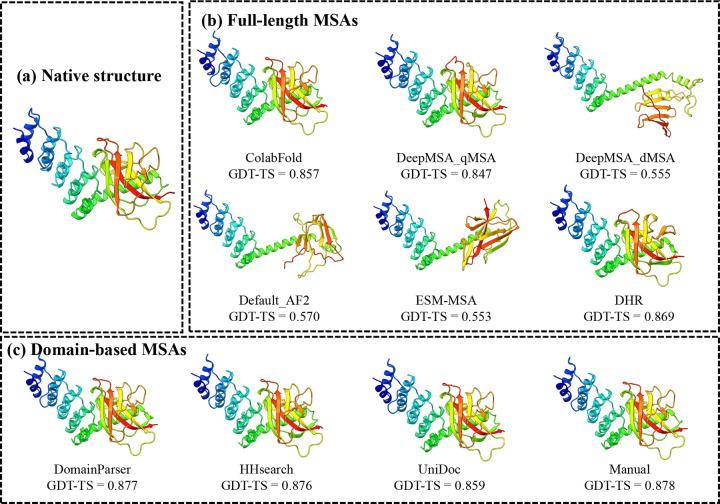
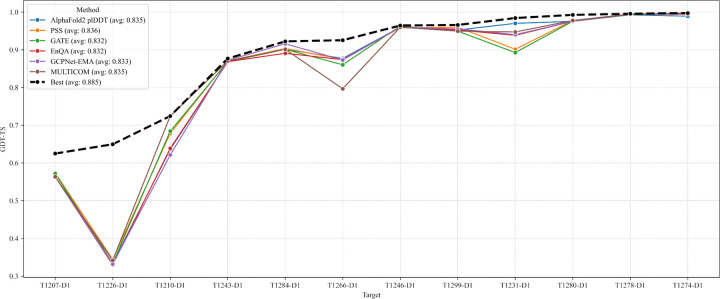
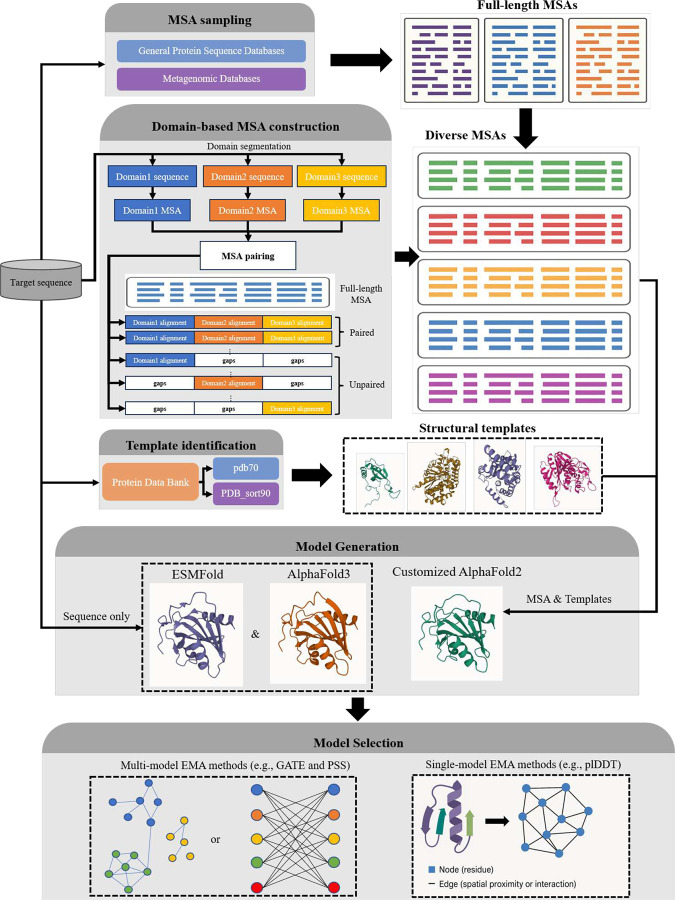
AlphaFold2 and AlphaFold3 have revolutionized protein structure prediction by enabling high-accuracy tertiary structure predictions for most single-chain proteins (monomers). However, obtaining high-quality predictions for some hard protein targets with shallow or noisy multiple sequence alignments (MSAs) and complicated multi-domain architectures remains challenging. Here, we present MULTICOM4, an integrative protein structure prediction system that uses diverse MSA generation, large-scale model sampling, and an ensemble model quality assessment (QA) strategy of combining individual QA methods to improve model generation and ranking of AlphaFold2 and AlphaFold3. In the 16th Critical Assessment of Techniques for Protein Structure Prediction (CASP16), our predictors built on MULTICOM4 ranked among the top performers out of 120 predictors in tertiary structure prediction and outperformed a standard AlphaFold3 predictor. The average TM-score of our best performing predictor MULTCOM's top-1 prediction for 84 CASP16 domain is 0.902. It achieved high accuracy (TM-score > 0.9) for 73.8% of the 84 domains and correct fold predictions (TM-score > 0.5) for 97.6% domains in terms of top-1 prediction. In terms of best-of-top-5 prediction, it predicted correct folds for all the domains. The results show that MSA engineering through the use of different protein sequence databases, alignment tools, and domain segmentation as well as extensive model sampling are the key to generate accurate and correct structural models. Additionally, using multiple complementary QA methods and model clustering can improve the robustness and reliability of model ranking.
Keywords: AlphaFold; deep learning; protein model quality assessment; protein structure prediction.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures
Similar articles
-
Boosting AlphaFold Protein Tertiary Structure Prediction through MSA Engineering and Extensive Model Sampling and Ranking in CASP16.bioRxiv [Preprint]. 2025 Jun 9:2025.06.06.658338. doi: 10.1101/2025.06.06.658338. bioRxiv. 2025. PMID: 40661500 Free PMC article. Preprint.
-
Improving AlphaFold2- and AlphaFold3-Based Protein Complex Structure Prediction With MULTICOM4 in CASP16.Proteins. 2025 Jun 2:10.1002/prot.26850. doi: 10.1002/prot.26850. Online ahead of print. Proteins. 2025. PMID: 40452318
-
Assessment of Protein Complex Predictions in CASP16: Are we making progress?bioRxiv [Preprint]. 2025 May 30:2025.05.29.656875. doi: 10.1101/2025.05.29.656875. bioRxiv. 2025. PMID: 40501681 Free PMC article. Preprint.
-
Unveiling the evolution of policies for enhancing protein structure predictions: A comprehensive analysis.Comput Biol Med. 2024 Sep;179:108815. doi: 10.1016/j.compbiomed.2024.108815. Epub 2024 Jul 11. Comput Biol Med. 2024. PMID: 38986287 Review.
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
References
-
- Senior A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
