

A PKCη missense mutation enhances Golgi-localized signaling and is associated with recessively inherited familial Alzheimer's disease
- PMID: 40591711
- PMCID: PMC12303577
- DOI: 10.1126/scisignal.adv0970
A PKCη missense mutation enhances Golgi-localized signaling and is associated with recessively inherited familial Alzheimer's disease
Abstract
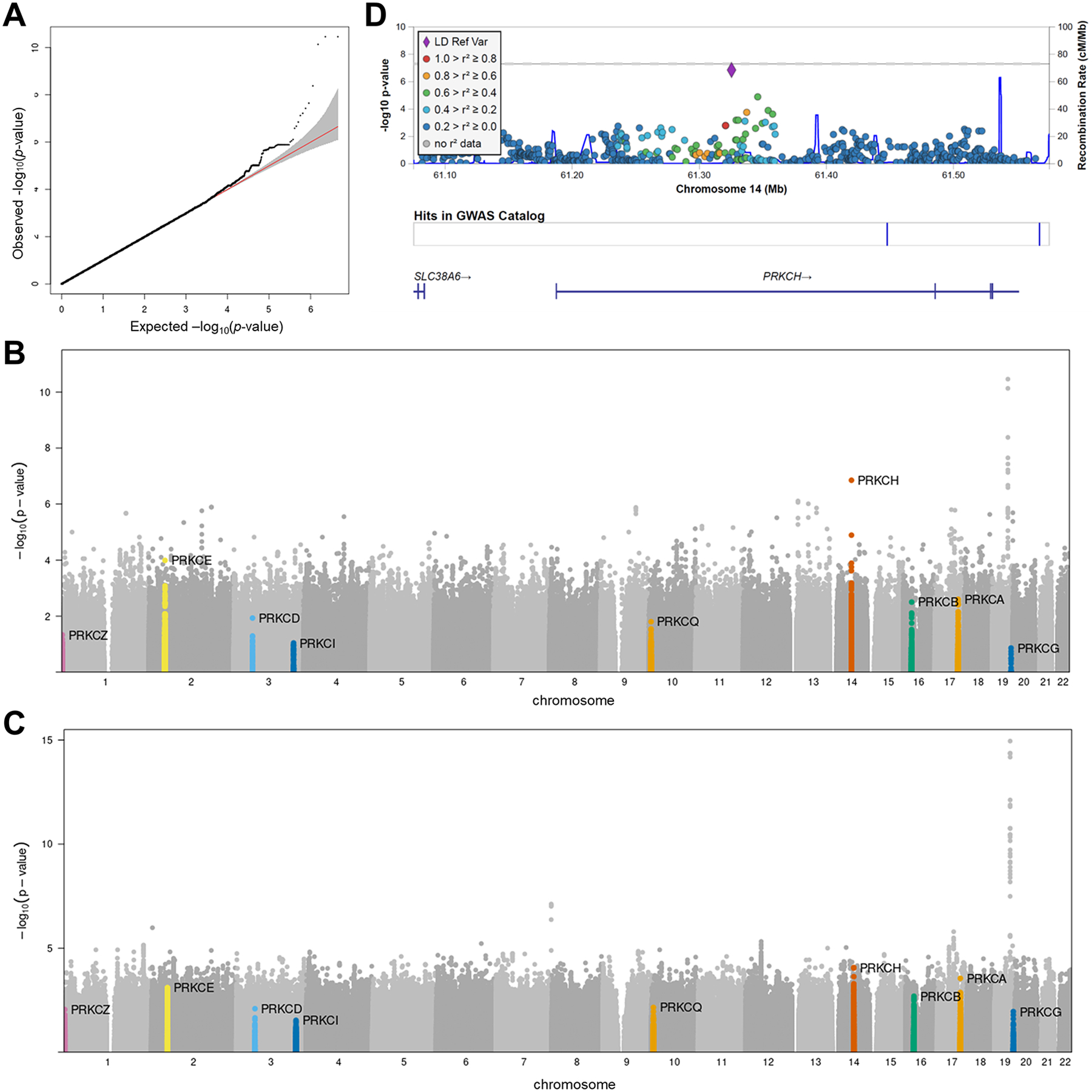
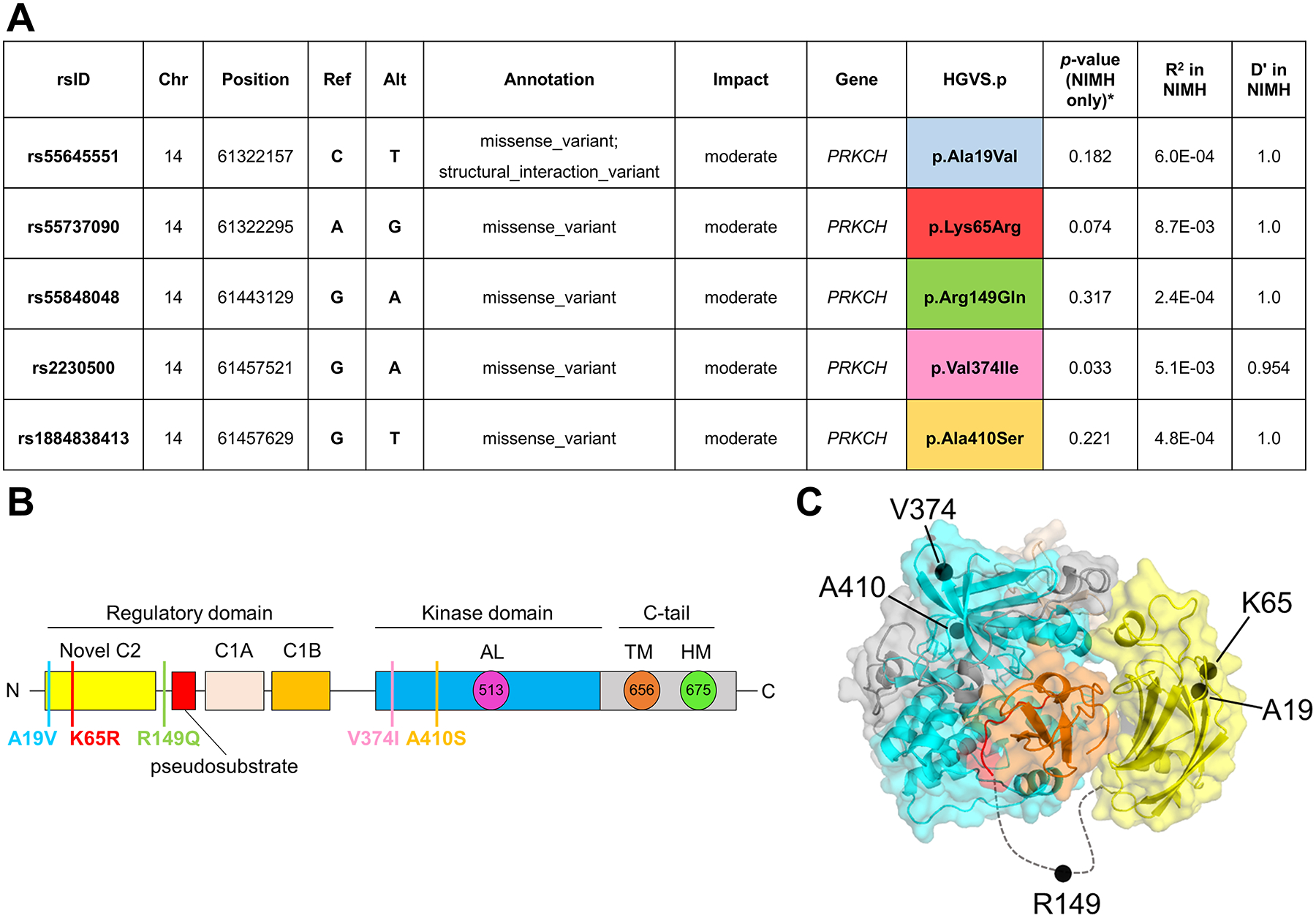
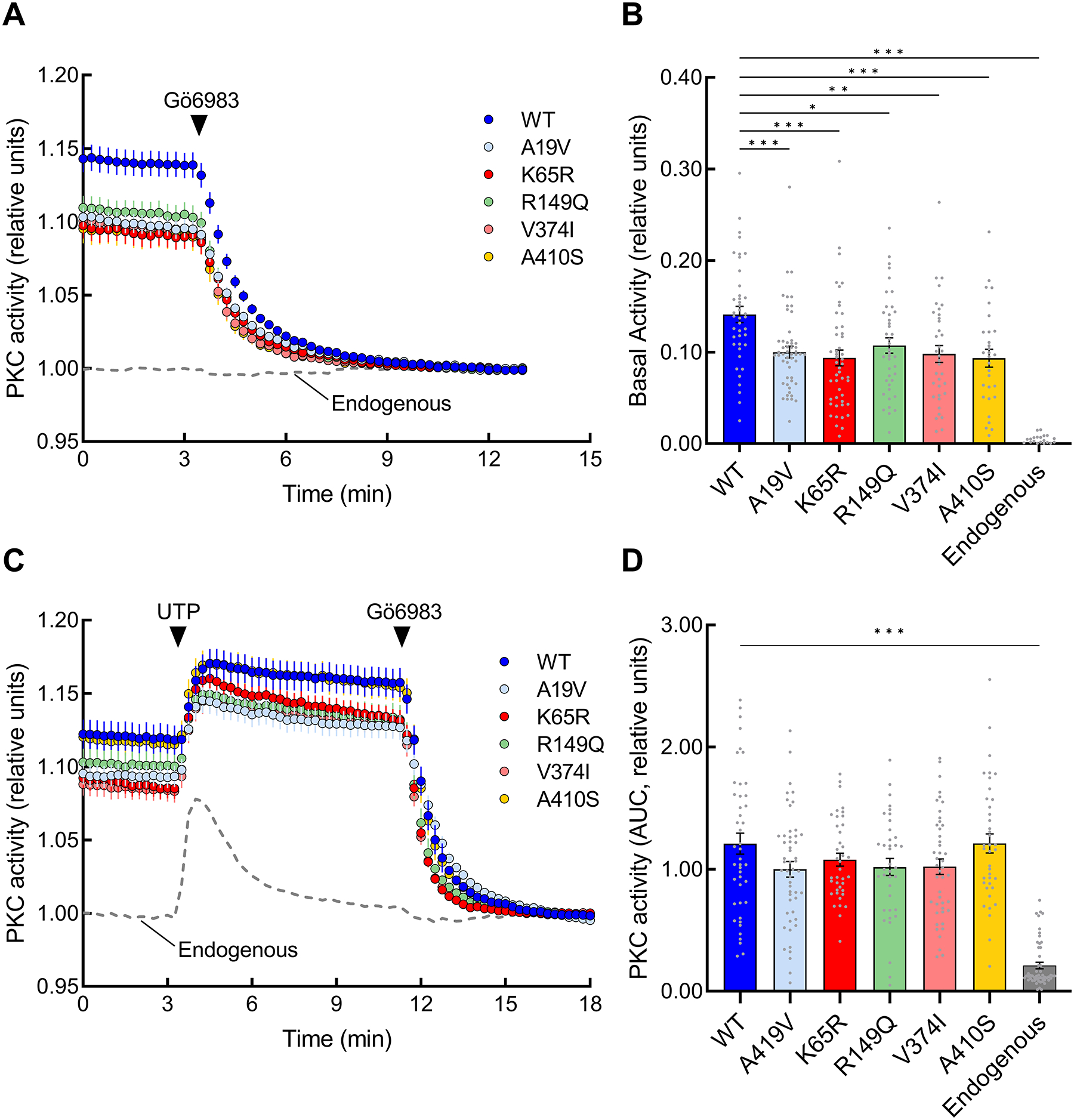
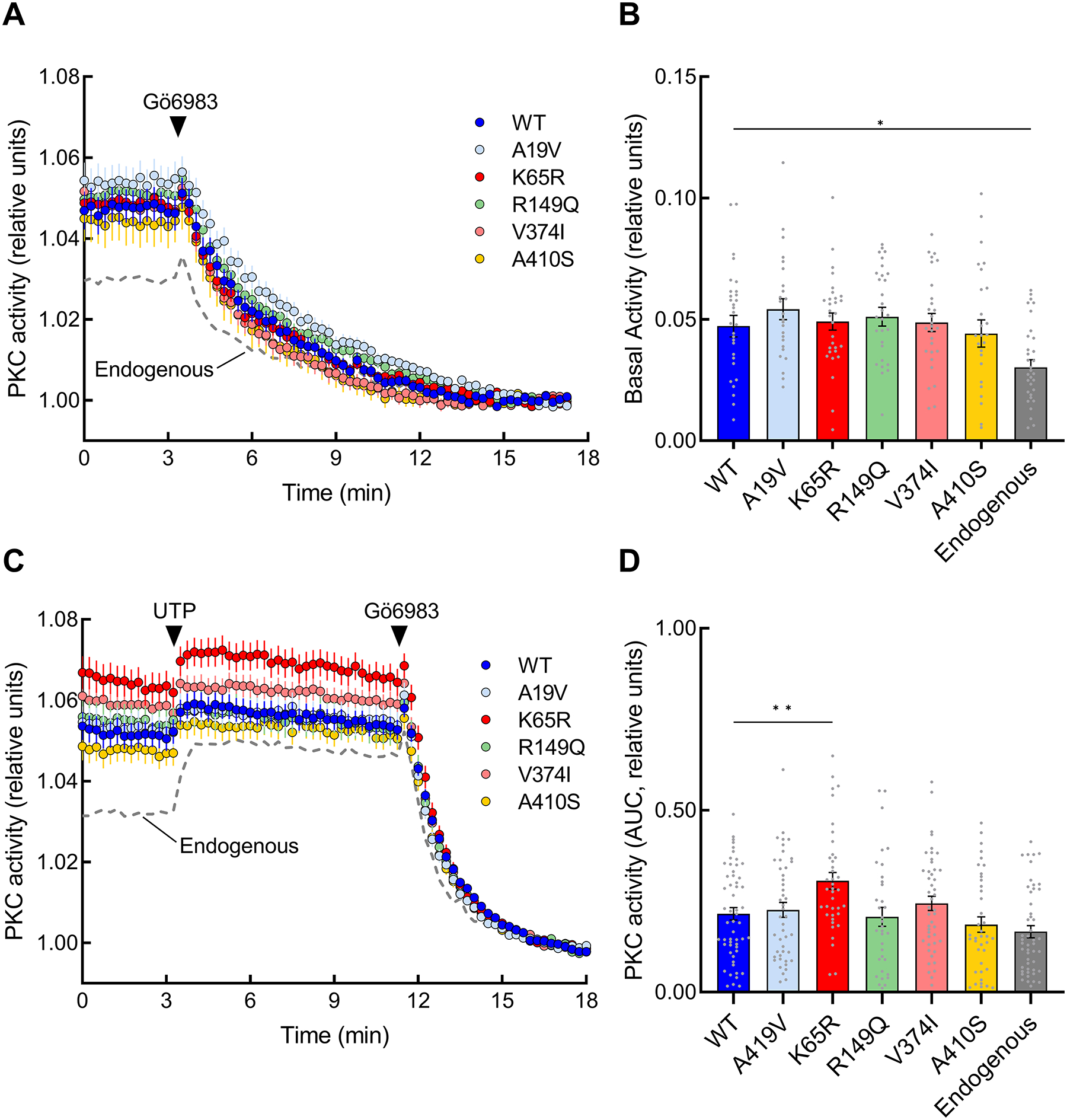
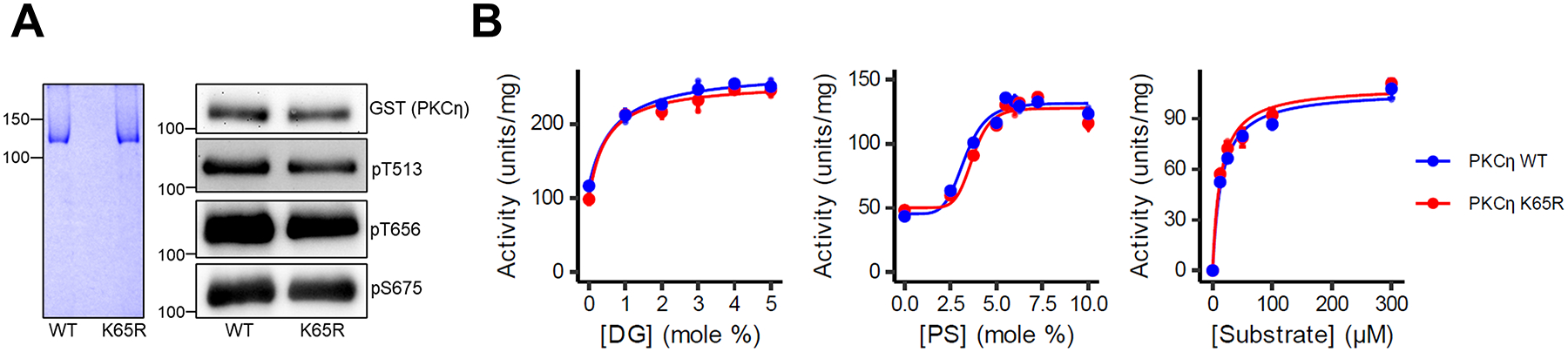
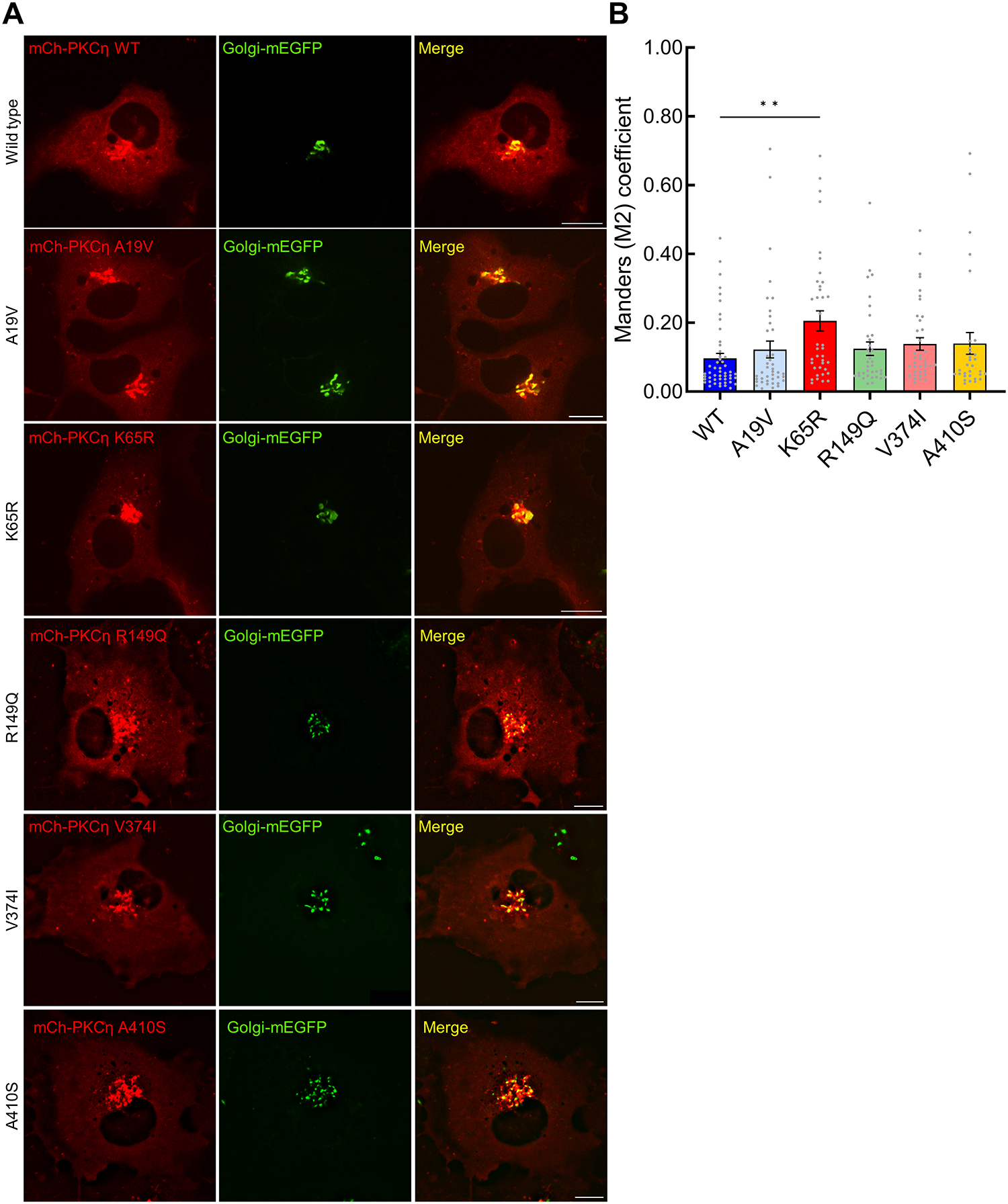
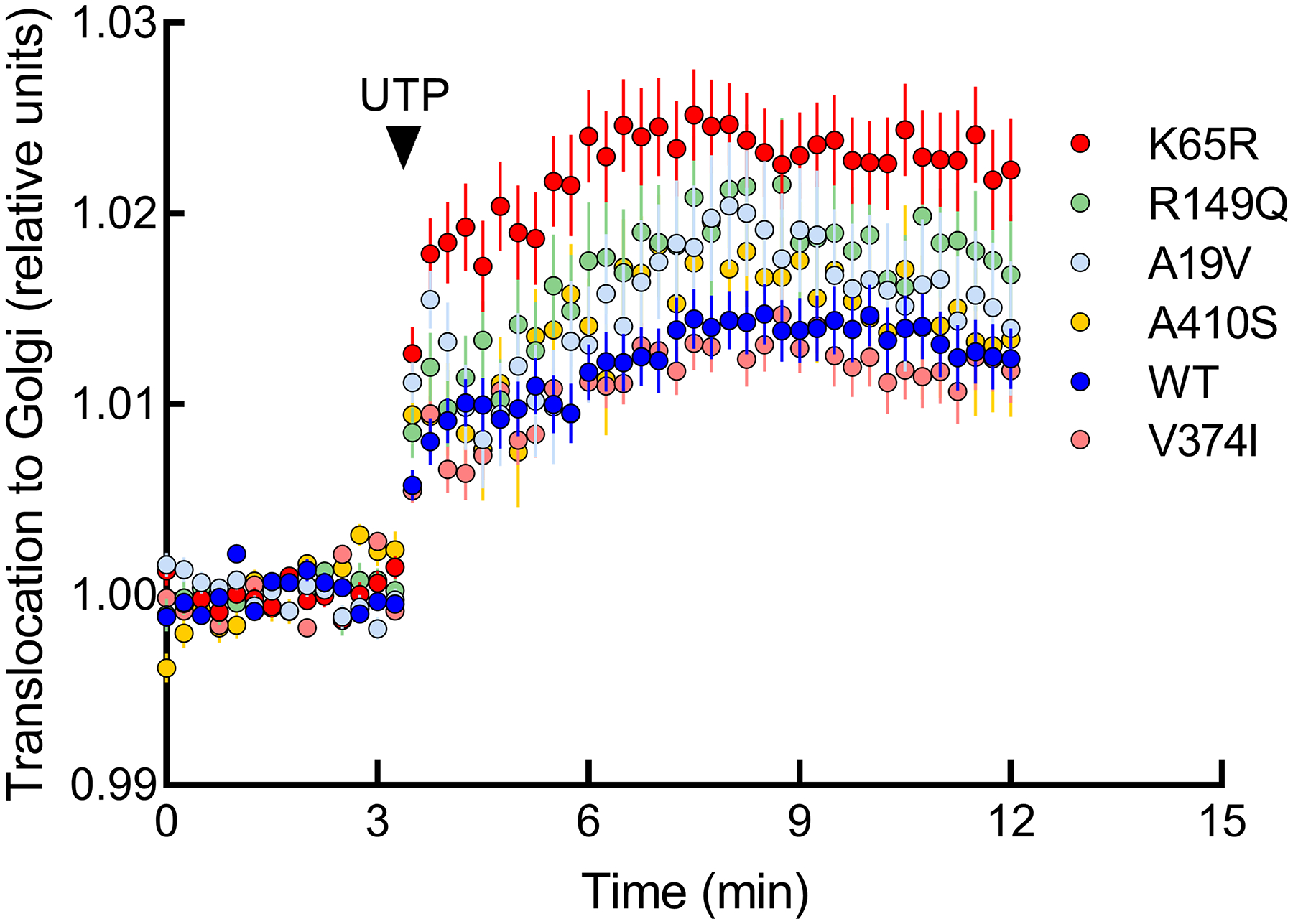
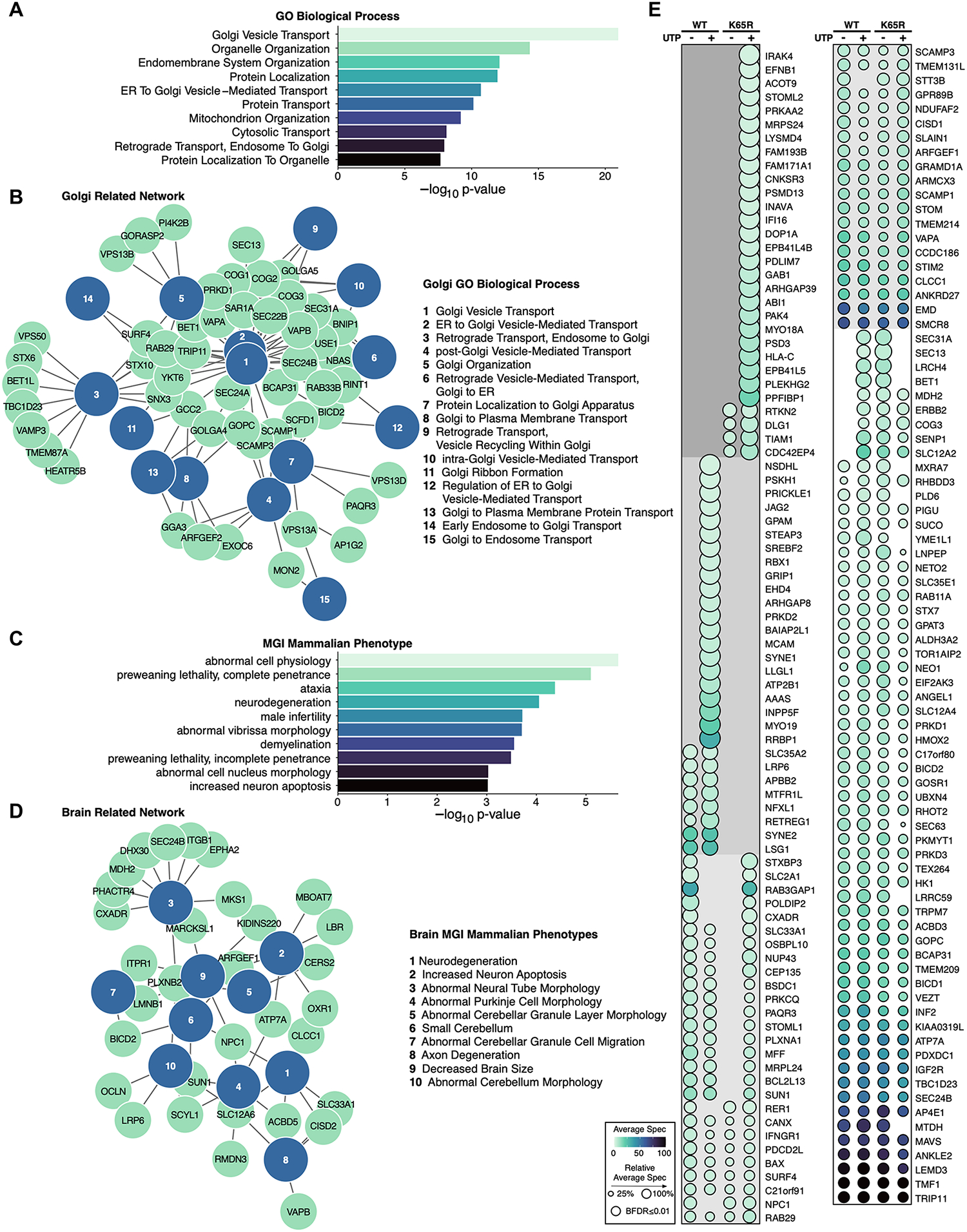
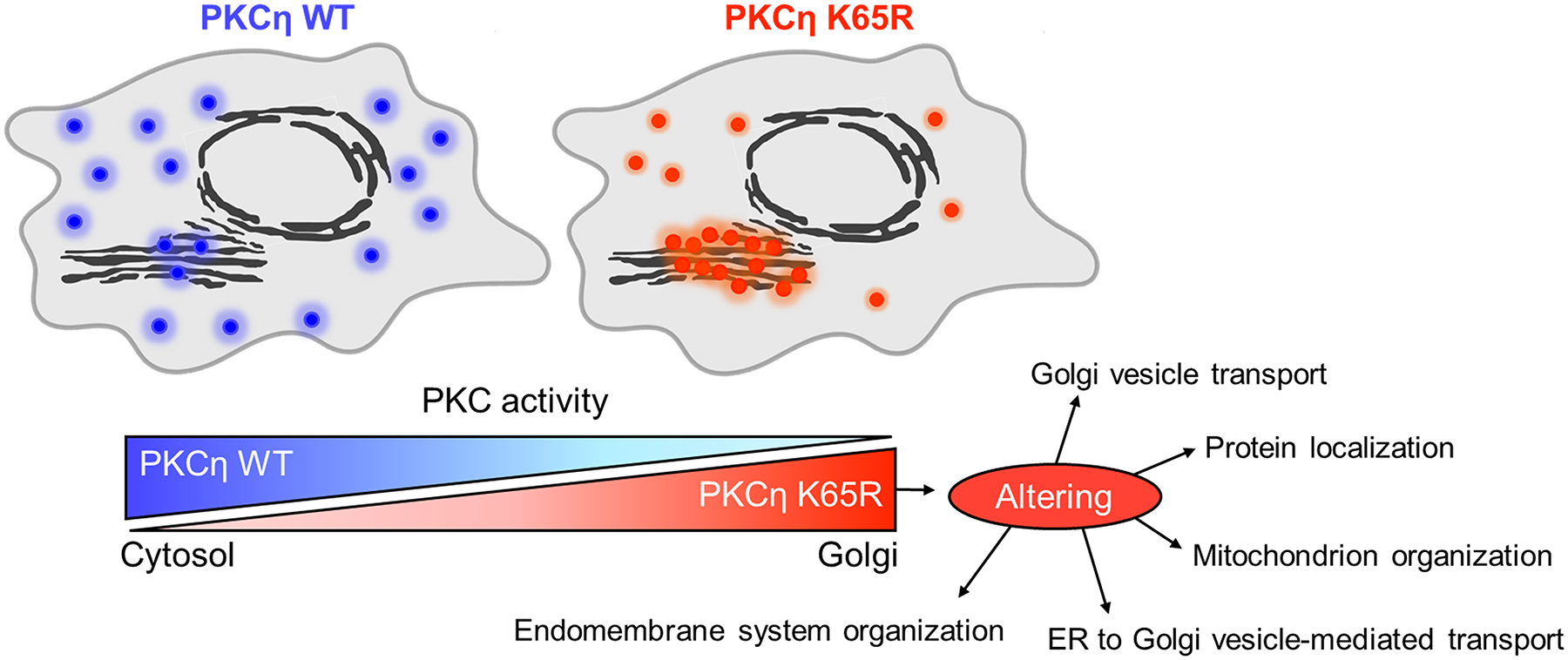
The identification of Alzheimer's disease (AD)-associated genomic variants has provided powerful insight into disease etiology. Genome-wide association studies (GWASs) of AD have successfully identified previously unidentified targets but have almost exclusively used additive genetic models. Here, we performed a family-based GWAS of a recessive inheritance model using whole-genome sequencing from families affected by AD. We found an association between AD risk and the variant rs7161410, which is located in an intron of the PRKCH gene encoding protein kinase C eta (PKCη). In addition, a rare PRKCH missense mutation, K65R, was in linkage disequilibrium with rs7161410 and was present in homozygous carriers of the rs7161410 risk allele. In vitro analysis revealed that the catalytic rate, lipid dependence, and peptide substrate binding of the purified variant were indistinguishable from those of the wild-type kinase. However, cellular studies revealed that the K65R PKCη variant had reduced cytosolic activity and, instead, enhanced localization and signaling at the Golgi. Moreover, the K65R variant had altered interaction networks in transfected cells, particularly with proteins involved in Golgi processes such as vesicle transport. In human brain tissue, the AD-associated recessive genotype of rs7161410 was associated with increased expression of PRKCH, particularly in the amygdala. This association of aberrant PKCη signaling with AD and the insight into how its function is altered may lead to previously unidentified therapeutic targets for prevention and treatment.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
