

Exploiting mitochondrial dysfunction to overcome BRAF inhibitor resistance in advanced melanoma: the role of disulfiram as a copper ionophore
- PMID: 40592836
- PMCID: PMC12216038
- DOI: 10.1038/s41419-025-07766-y
Exploiting mitochondrial dysfunction to overcome BRAF inhibitor resistance in advanced melanoma: the role of disulfiram as a copper ionophore
Abstract
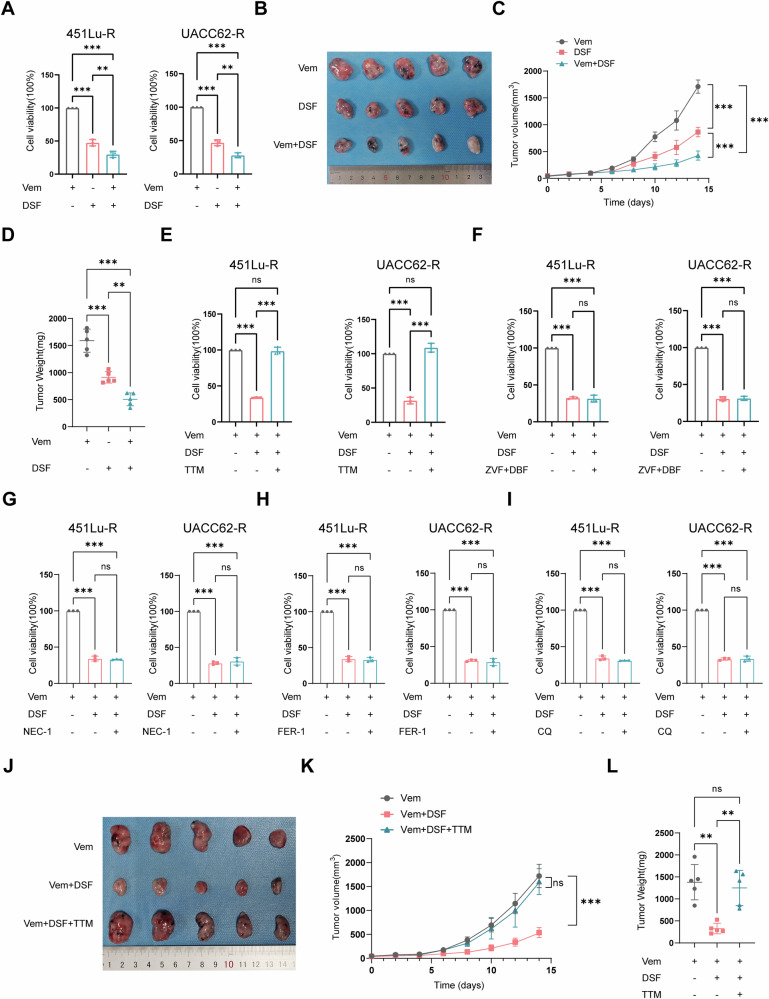
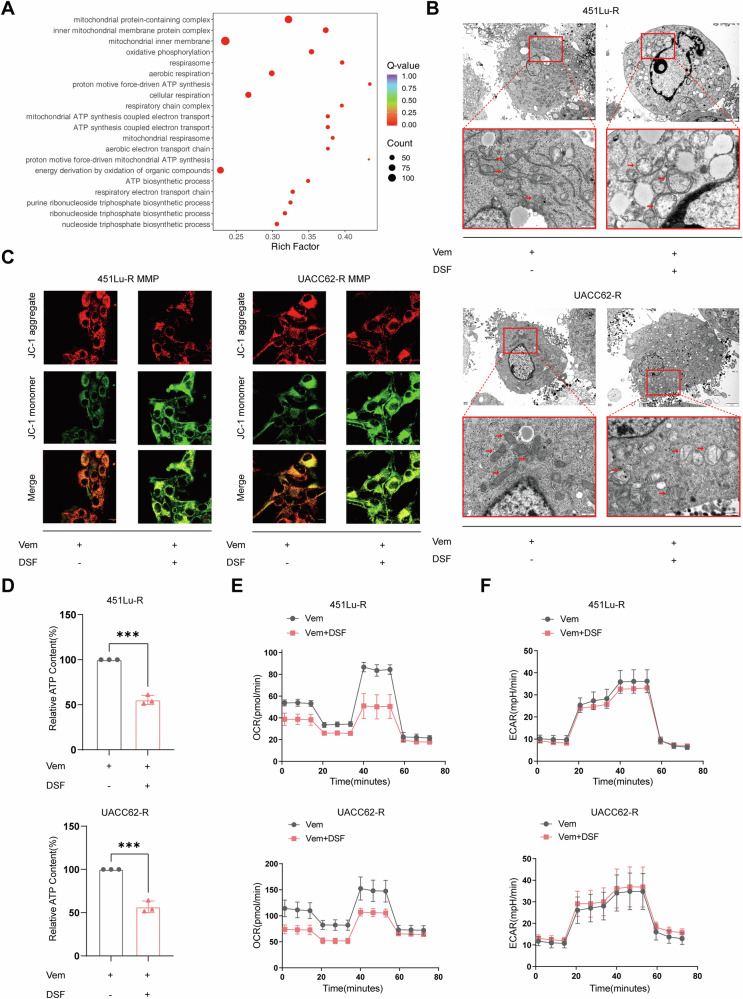
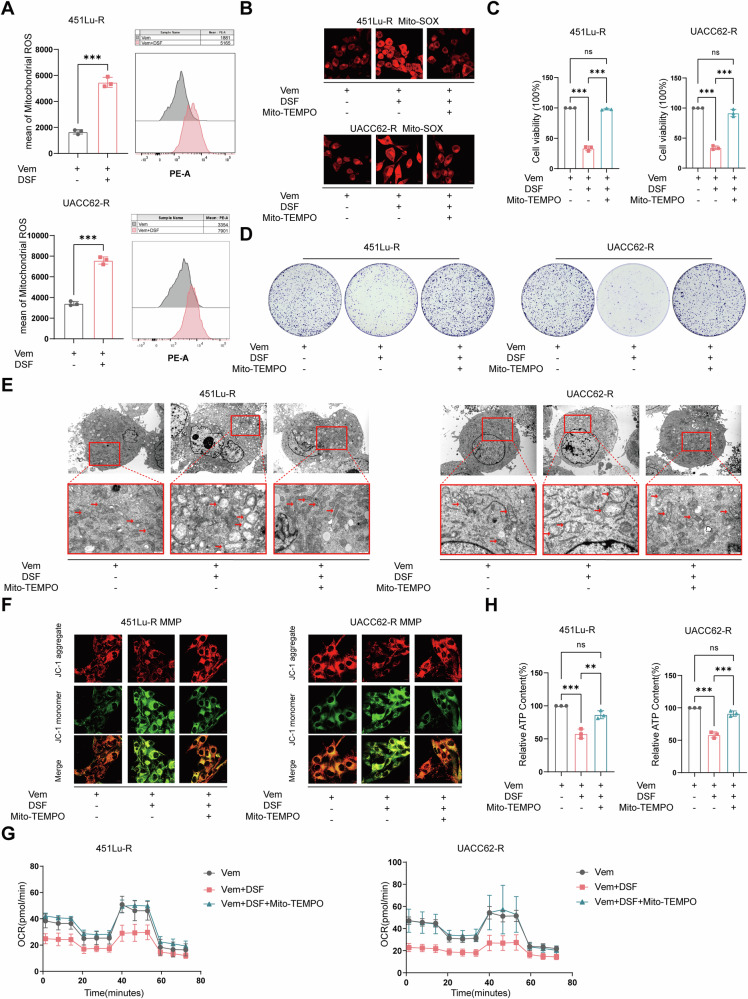
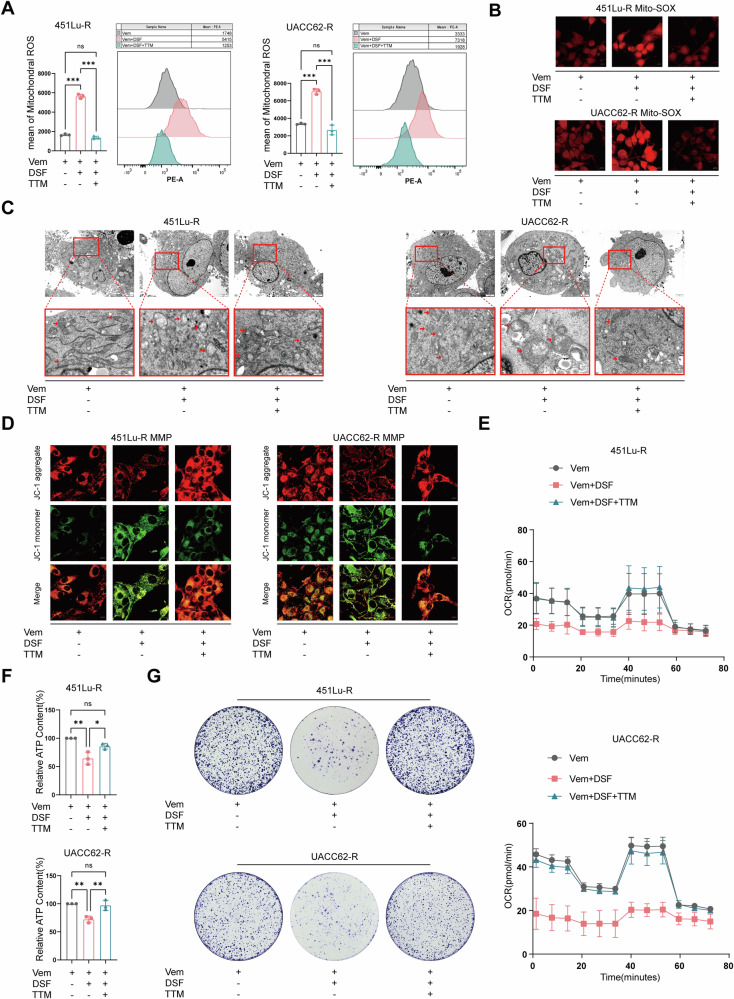
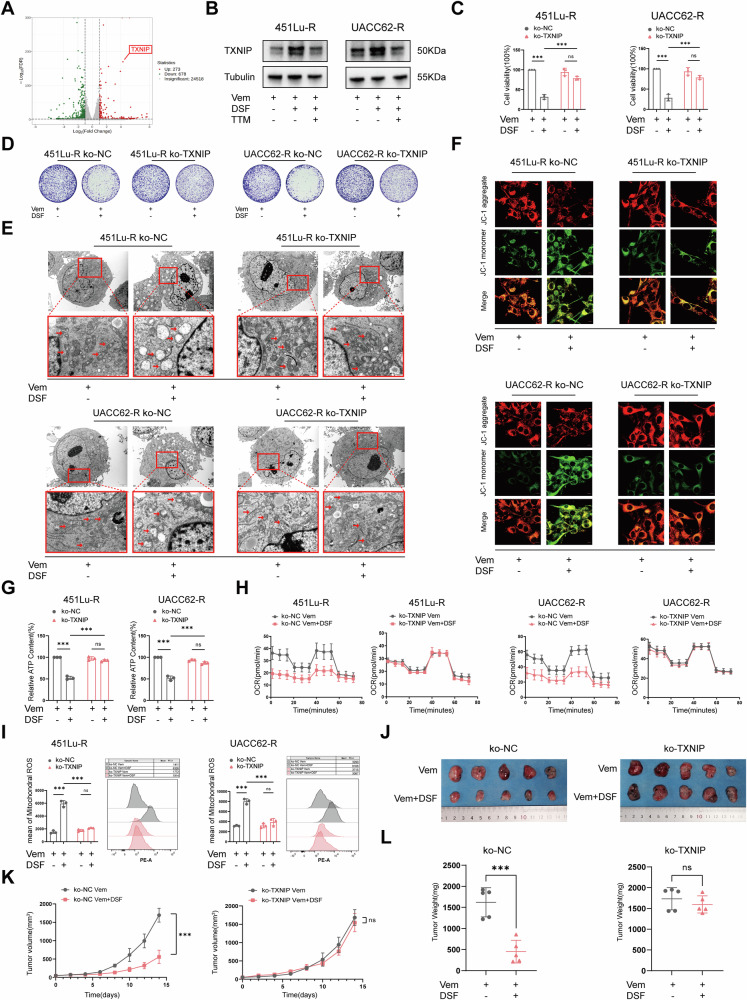
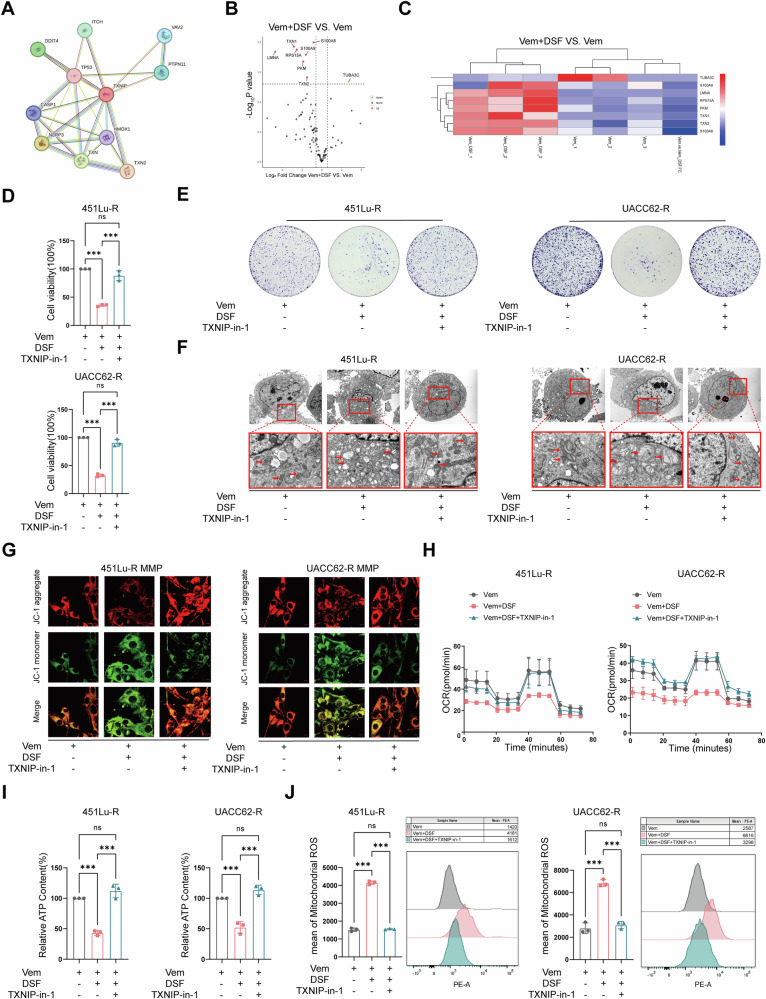
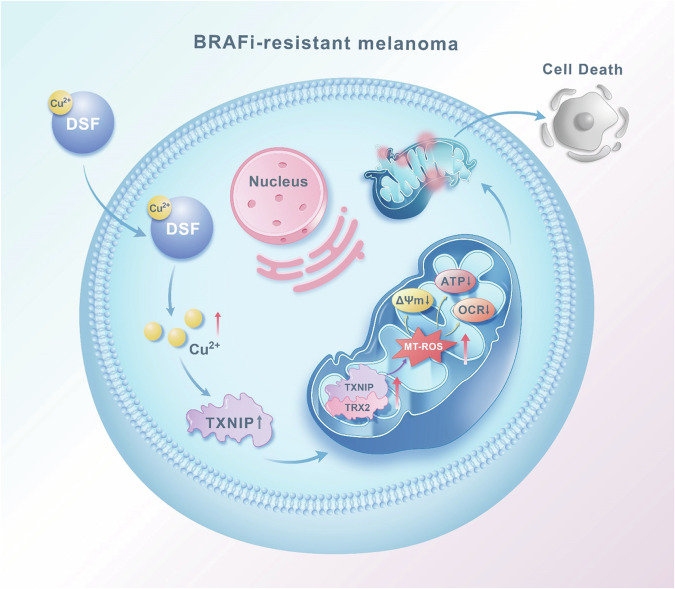
Resistance to targeted therapies poses a significant challenge in advanced melanoma with BRAF mutations. Even with a BRAF + MEK inhibitor combination, about 70% of patients experience disease progression within two years, highlighting the need for novel strategies beyond MAPK signaling inhibition. This study investigates whether mitochondrial dysfunction induced by the copper ionophore disulfiram (DSF) can effectively counteract resistance to BRAF inhibitors. We established two BRAF inhibitor (BRAFi)-resistant melanoma cell lines using BRAF mutant 451Lu and UACC62. In vivo experiments were conducted using subcutaneous implantation in nude mice. Cell viability and colony formation assays assessed treatment efficacy, while mitochondrial morphology was evaluated via transmission electron microscopy. Mitochondrial respiration was measured using a Seahorse metabolic analyzer, and oxidative stress was assessed through flow cytometry and confocal microscopy. RNA sequencing identified downstream factors regulated by intracellular copper levels, and the CRISPR-Cas9 system was used to knock out candidate genes in BRAFi-resistant cells for mechanistic validation. We provided evidence that DSF induced cell death in BRAFi-resistant melanoma in a copper-dependent manner, severely impairing mitochondrial structure and function through increased oxidative stress. RNA-seq and immunoblotting revealed that thioredoxin-interacting protein (TXNIP) expression significantly increased in response to DSF. TXNIP knockout reduced DSF-induced cytotoxicity by mitigating oxidative stress. These findings were supported by in vivo experiments. Furthermore, we demonstrated that the oxidative damage mediated by TXNIP involved its interaction with thioredoxin 2 (TRX2). In conclusion, targeting mitochondrial function with disulfiram effectively inhibits BRAFi-resistant melanoma cells, independent of MAPK signaling blockage. These results point to the potential of combining disulfiram with BRAF inhibitors as a promising strategy to overcome BRAFi resistance.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics approval: The study was approved by the Animal Protection and Utilization Committee of the animal facility at the Fourth Military Medical University. All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animal by International Committees. Every effort was made to minimize the numbers and suffering of the included animals.
Figures
Similar articles
-
Downregulation of lncRNA TSLNC8 promotes melanoma resistance to BRAF inhibitor PLX4720 through binding with PP1α to re-activate MAPK signaling.J Cancer Res Clin Oncol. 2021 Mar;147(3):767-777. doi: 10.1007/s00432-020-03484-4. Epub 2021 Jan 3. J Cancer Res Clin Oncol. 2021. PMID: 33389075 Free PMC article.
-
Protein phosphatase 6 activates NF-κB to confer sensitivity to MAPK pathway inhibitors in KRAS- and BRAF-mutant cancer cells.Sci Signal. 2024 May 14;17(836):eadd5073. doi: 10.1126/scisignal.add5073. Epub 2024 May 14. Sci Signal. 2024. PMID: 38743809 Free PMC article.
-
FAK inhibition combined with the RAF-MEK clamp avutometinib overcomes resistance to targeted and immune therapies in BRAF V600E melanoma.Cancer Cell. 2025 Mar 10;43(3):428-445.e6. doi: 10.1016/j.ccell.2025.02.001. Epub 2025 Feb 27. Cancer Cell. 2025. PMID: 40020669
-
Improved outcomes in women with BRAF-mutant melanoma treated with BRAF/MEK-targeted therapy across randomized clinical trials. A systematic review and meta-analysis.Semin Oncol. 2023 Feb-Apr;50(1-2):34-39. doi: 10.1053/j.seminoncol.2023.03.003. Epub 2023 Mar 21. Semin Oncol. 2023. PMID: 36967333
-
Immunohistochemistry as a reliable method for detection of BRAF-V600E mutation in melanoma: a systematic review and meta-analysis of current published literature.J Surg Res. 2016 Jun 15;203(2):407-15. doi: 10.1016/j.jss.2016.04.029. Epub 2016 Apr 23. J Surg Res. 2016. PMID: 27363650
References
-
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. - PubMed
-
- Larkin J, Lao CD, Urba WJ, Mcdermott DF, Horak C, Jiang J, et al. Efficacy and safety of nivolumab in patients with BRAF V600 mutant and BRAF wild-type advanced melanoma: a pooled analysis of 4 clinical trials. JAMA Oncol. 2015;1:433–40. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
