

Prosit-XL: enhanced cross-linked peptide identification by fragment intensity prediction to study protein interactions and structures
- PMID: 40592844
- PMCID: PMC12214610
- DOI: 10.1038/s41467-025-61203-4
Prosit-XL: enhanced cross-linked peptide identification by fragment intensity prediction to study protein interactions and structures
Abstract
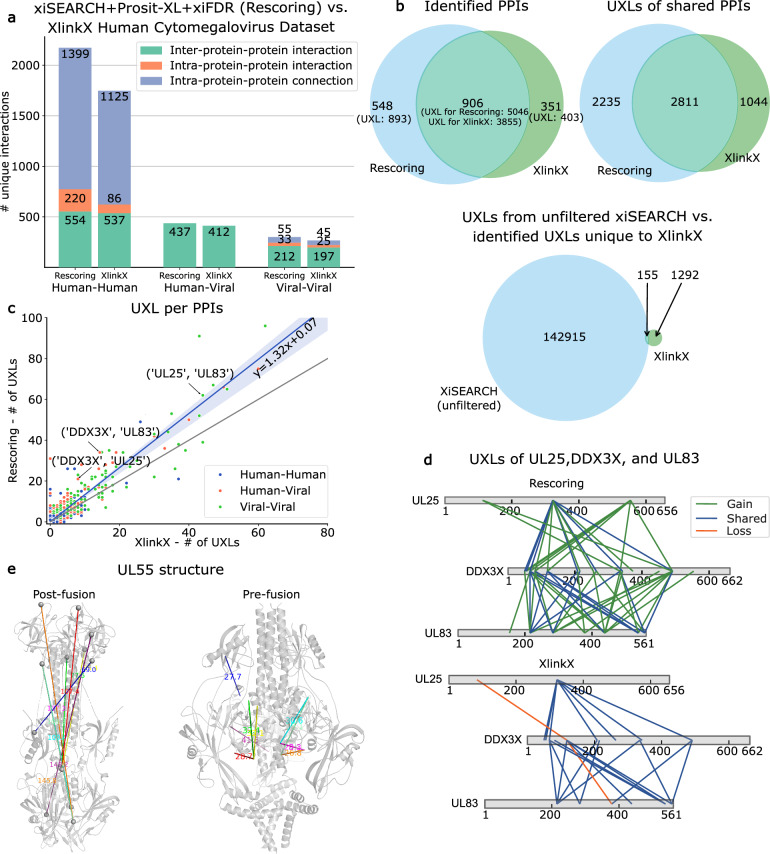
It has been shown that integrating peptide property predictions such as fragment intensity into the scoring process of peptide spectrum match can greatly increase the number of confidently identified peptides compared to using traditional scoring methods. Here, we introduce Prosit-XL, a robust and accurate fragment intensity predictor covering the cleavable (DSSO/DSBU) and non-cleavable cross-linkers (DSS/BS3), achieving high accuracy on various holdout sets with consistent performance on external datasets without fine-tuning. Due to the complex nature of false positives in XL-MS, an approach to data-driven rescoring was developed that benefits from Prosit-XL's predictions while limiting the overestimation of the false discovery rate (FDR). After validating this approach using two ground truth datasets consisting of synthetic peptides and proteins, we applied Prosit-XL on a proteome-scale dataset, demonstrating an up to ~3.4-fold improvement in PPI discovery compared to classic approaches. Finally, Prosit-XL was used to increase the coverage and depth of a spatially resolved interactome map of intact human cytomegalovirus virions, leading to the discovery of previously unobserved interactions between human and cytomegalovirus proteins.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.W. is a founder and shareholder of MSAID GmbH with no operational role and member of the scientific advisory board of Momentum Biotechnologies. The remaining authors declare no competing interests.
Figures
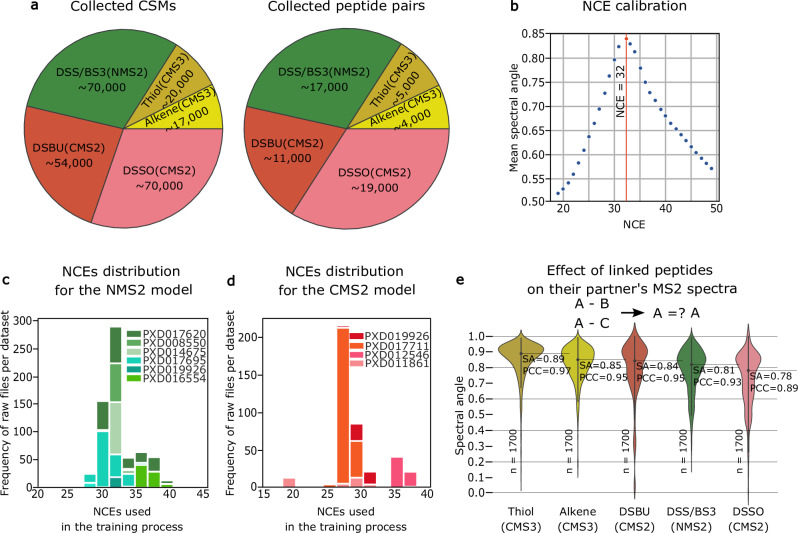
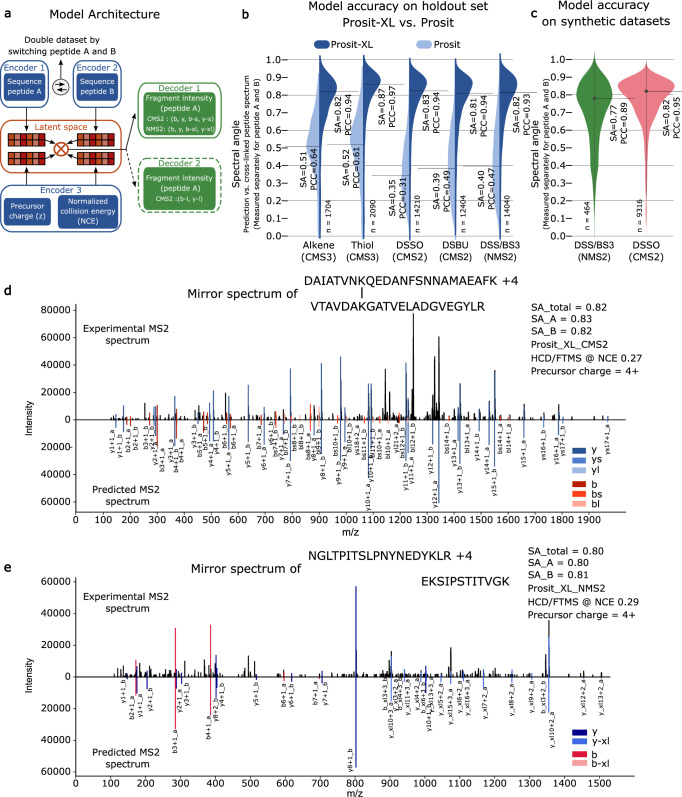
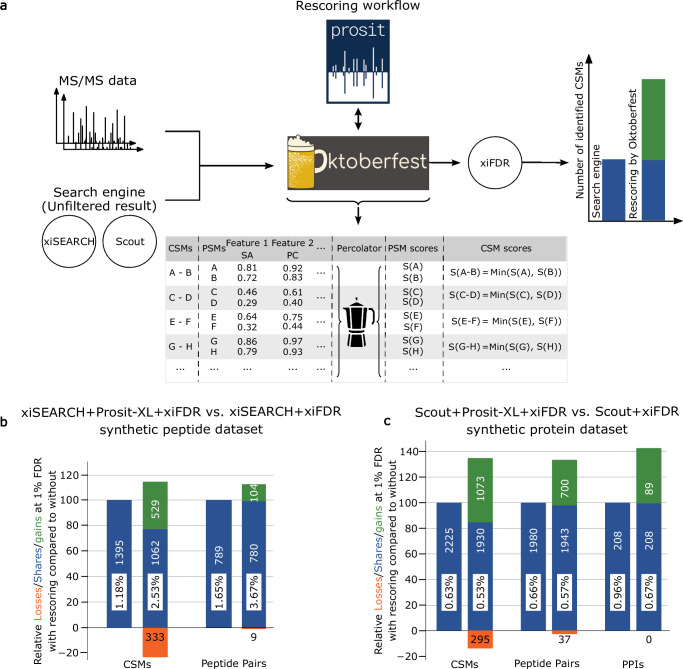
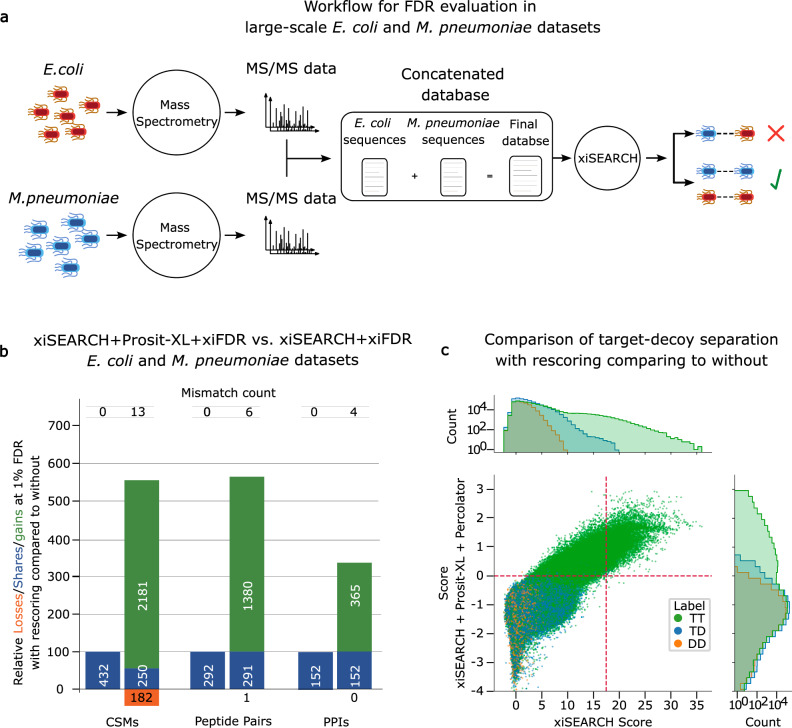
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
