

Spatially resolved C1QC+ macrophage-CD4+ T cell niche in colorectal cancer microenvironment: implications for immunotherapy response
- PMID: 40593467
- PMCID: PMC12219098
- DOI: 10.1038/s41421-025-00811-2
Spatially resolved C1QC+ macrophage-CD4+ T cell niche in colorectal cancer microenvironment: implications for immunotherapy response
Abstract
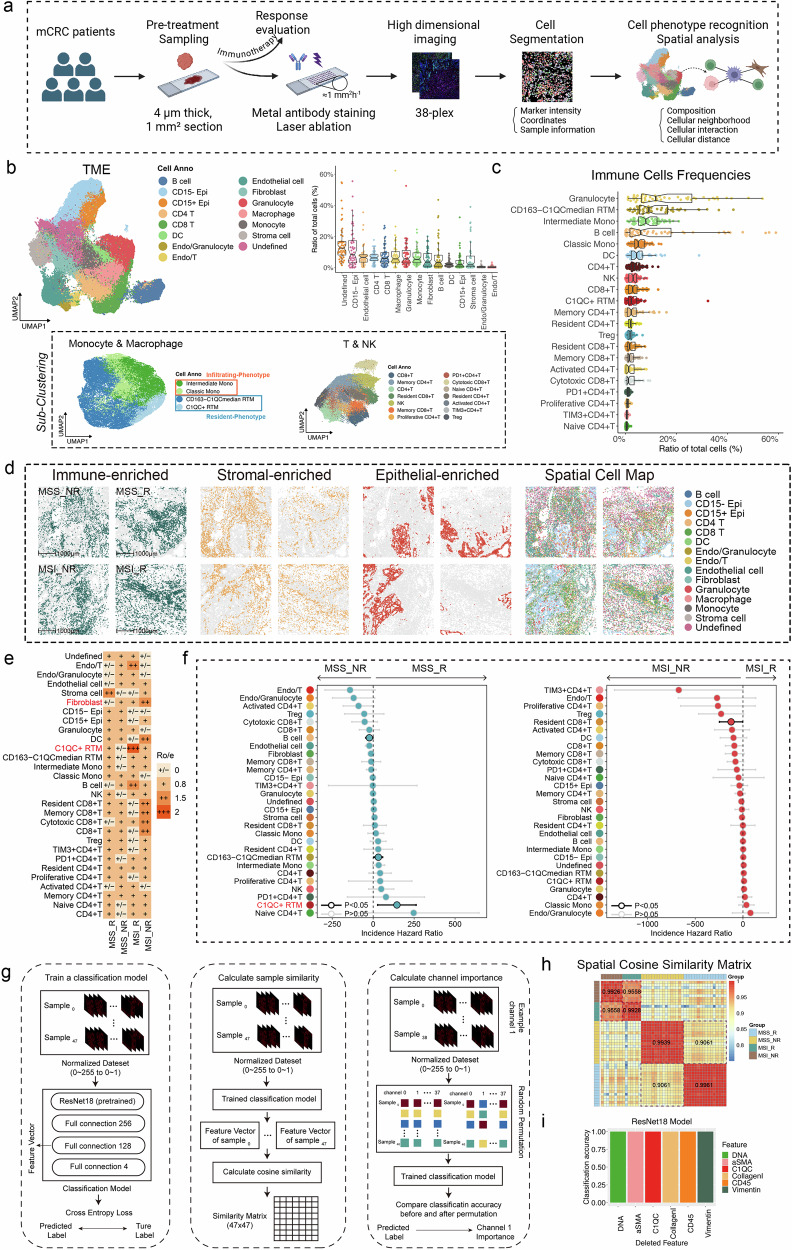
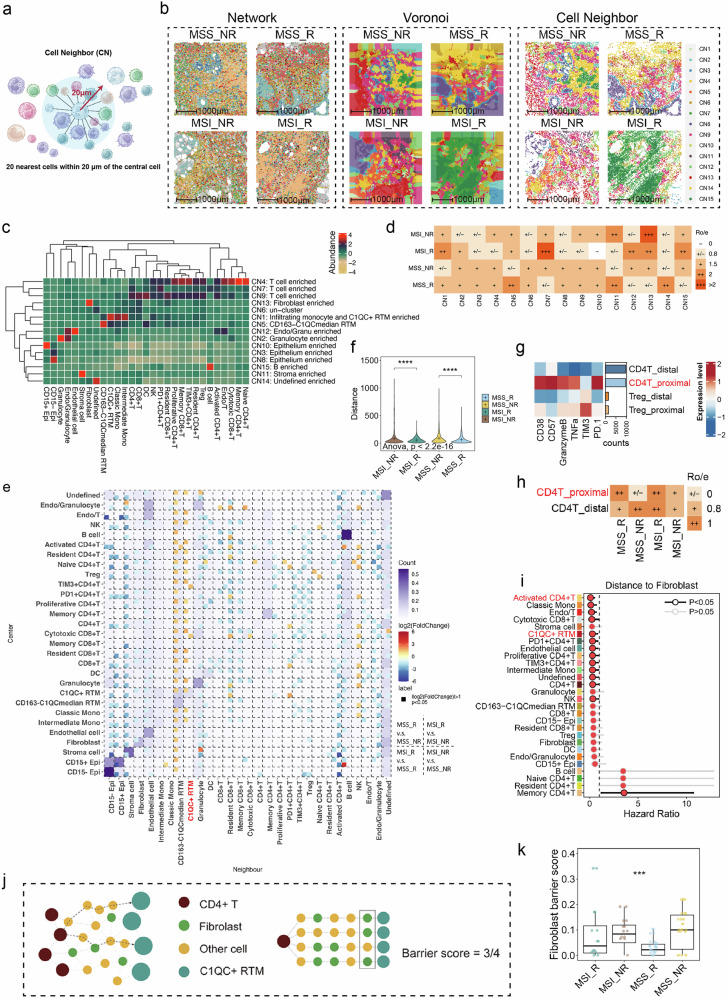
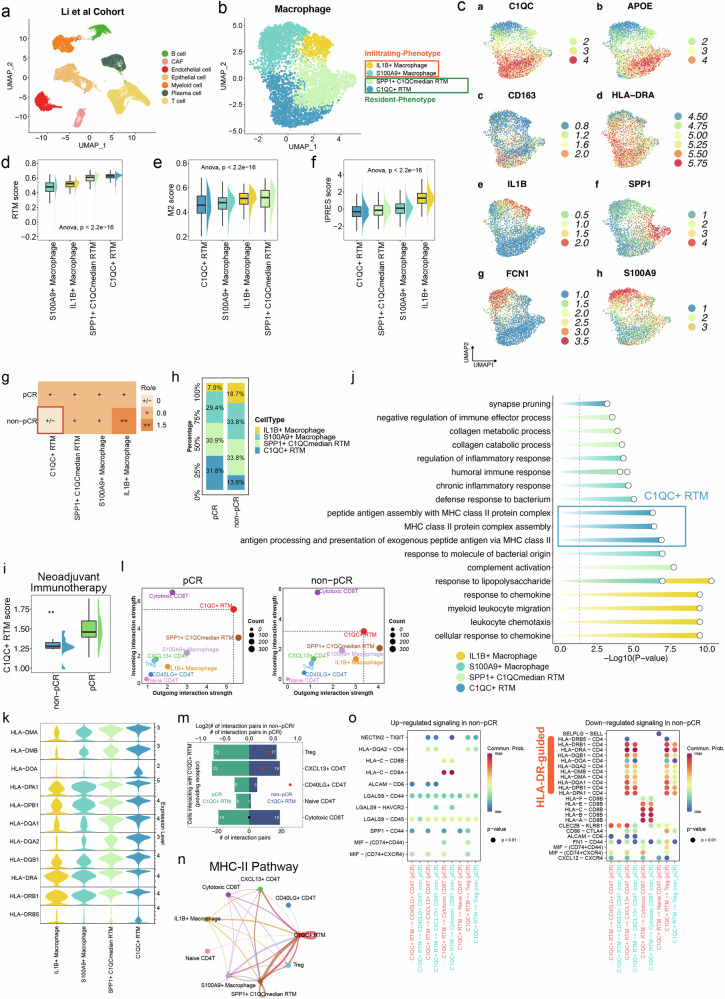
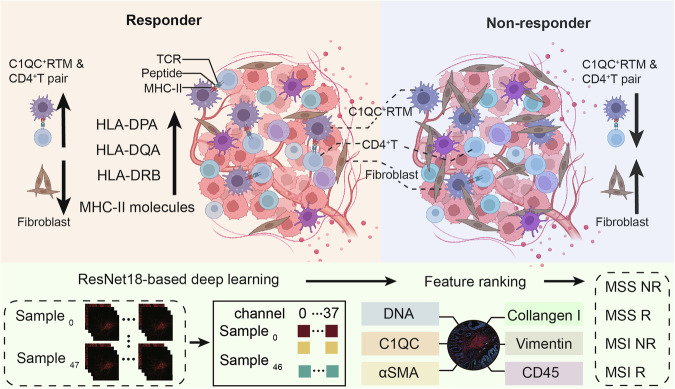
Colorectal cancer (CRC), including both microsatellite instability (MSI) and microsatellite stability (MSS) subtypes, frequently exhibits intrinsic resistance to immunotherapy. However, the spatial tumor microenvironment (TME) and its role in distinguishing immunotherapy responders from non-responders remain poorly understood. In this study, spatial multiomics, including imaging mass cytometry (n = 50 in-house), spatial proteomics (n = 50 in-house), and spatial transcriptomics (n = 9 in-house), were employed to elucidate the spatial TME of metastatic CRC (mCRC) patients receiving immunotherapy. These methodologies were integrated with single-cell RNA sequencing (scRNA-seq), bulk RNA-seq, and bulk proteomics for comprehensive analysis and validation. A spatial immune atlas containing 314,774 cells was constructed. We found that C1QC+ resident tissue macrophages (RTMs) were more abundant in responders regardless of microsatellite status. Co-localization of C1QC+ RTMs with CD4+ T cells was observed in responders, and MHC-II expression facilitated their interaction. In contrast, cancer-associated fibroblasts inhibited this interaction in non-responders. Moreover, whole genome screening identified key genes involved in antigen presentation in C1QC+ RTMs. Hence, our study highlights the importance of spatial immune mapping in revealing the complex spatial topology of CRC and corresponding immunotherapy response.
© 2025. The Author(s).
Conflict of interest statement
Conflict of interest: The authors declare no competing interests.
Figures




References
Grants and funding
- 82101830/National Natural Science Foundation of China (National Science Foundation of China)
- 82473471/National Natural Science Foundation of China (National Science Foundation of China)
- 82202875/National Natural Science Foundation of China (National Science Foundation of China)
- 82303850/National Natural Science Foundation of China (National Science Foundation of China)
- 82102817/National Natural Science Foundation of China (National Science Foundation of China)
- 92259201/National Natural Science Foundation of China (National Science Foundation of China)
- LY23H160013/Natural Science Foundation of Zhejiang Province (Zhejiang Provincial Natural Science Foundation)
- LQ23H160041/Natural Science Foundation of Zhejiang Province (Zhejiang Provincial Natural Science Foundation)
- LQ23H160029/Natural Science Foundation of Zhejiang Province (Zhejiang Provincial Natural Science Foundation)
- LQ24H160017/Natural Science Foundation of Zhejiang Province (Zhejiang Provincial Natural Science Foundation)
- 2023C03061/Natural Science Foundation of Zhejiang Province (Zhejiang Provincial Natural Science Foundation)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
