

p66Shc deletion confers apoptotic resistance to loss of EGFR-ERK signalling in neural stem cells
- PMID: 40593476
- PMCID: PMC12217751
- DOI: 10.1038/s41419-025-07778-8
p66Shc deletion confers apoptotic resistance to loss of EGFR-ERK signalling in neural stem cells
Abstract
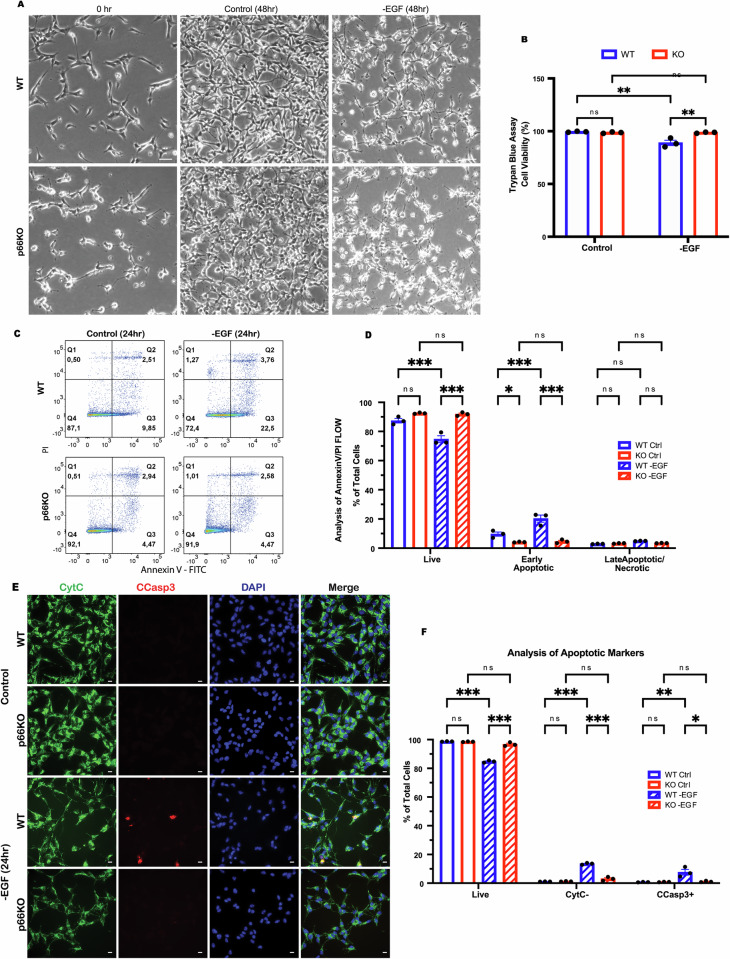
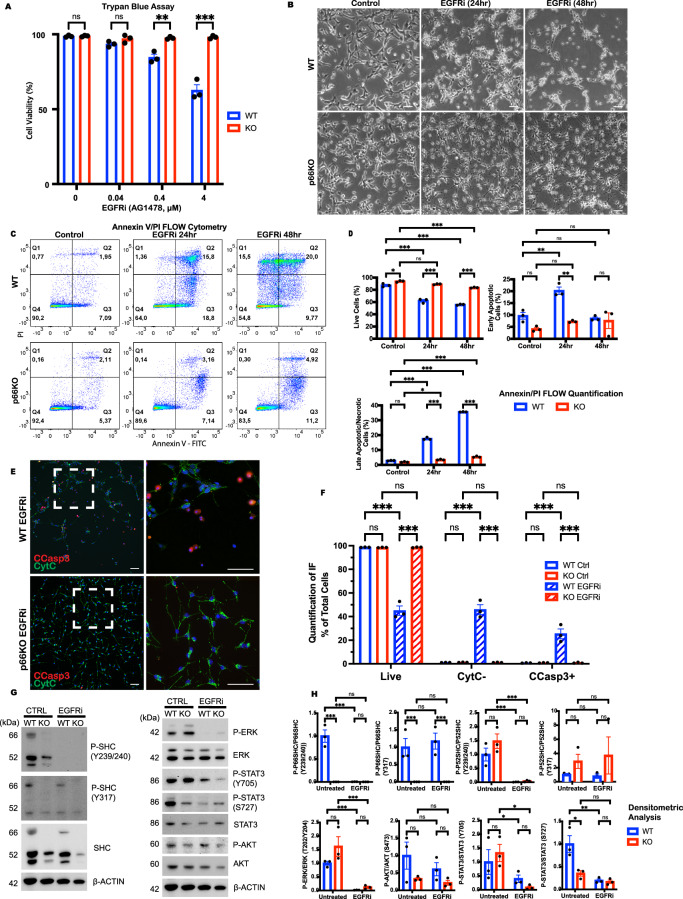
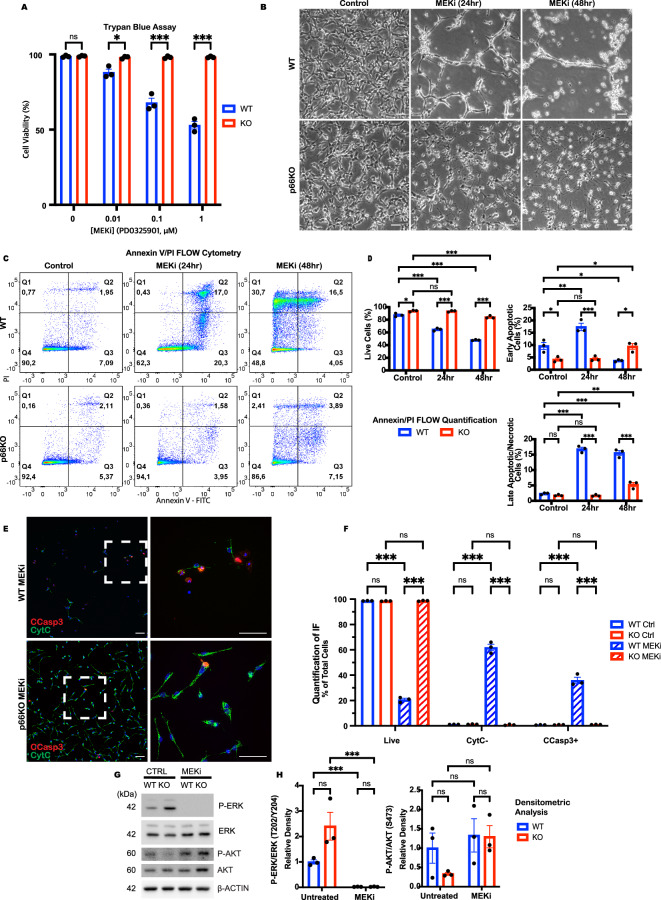
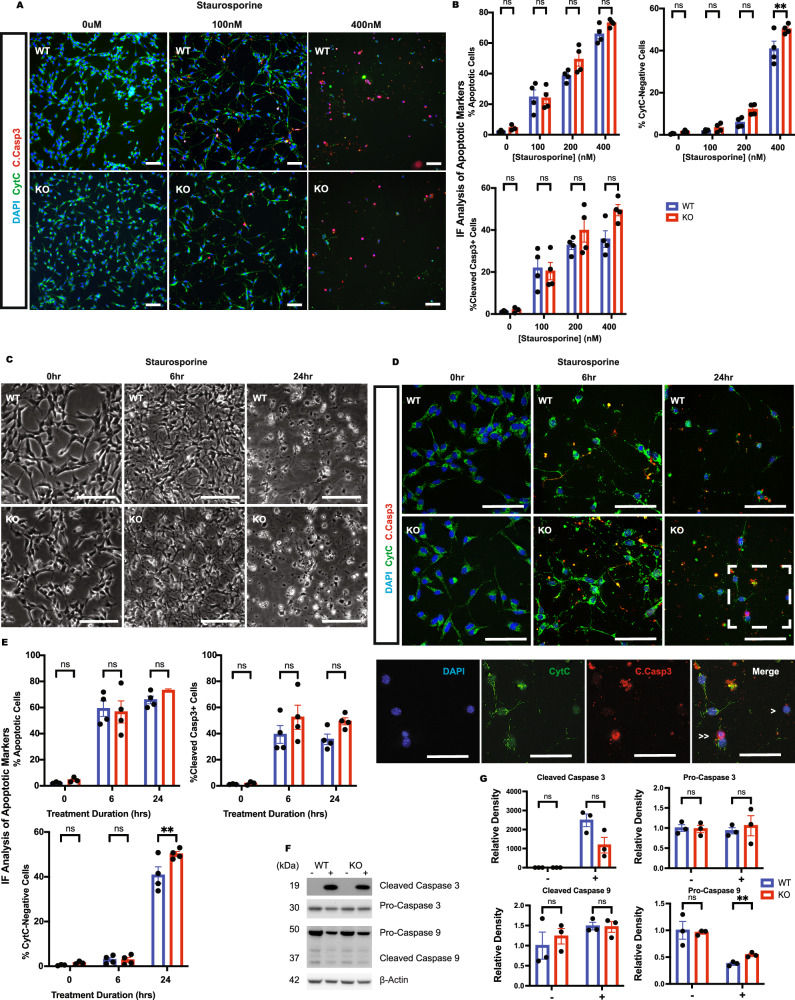
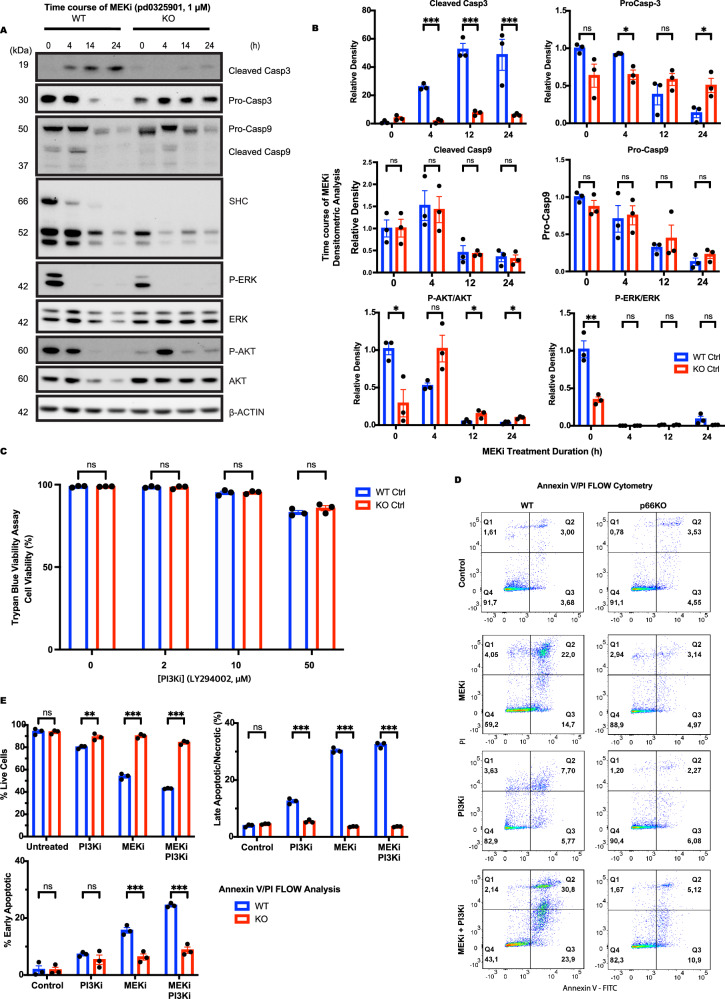
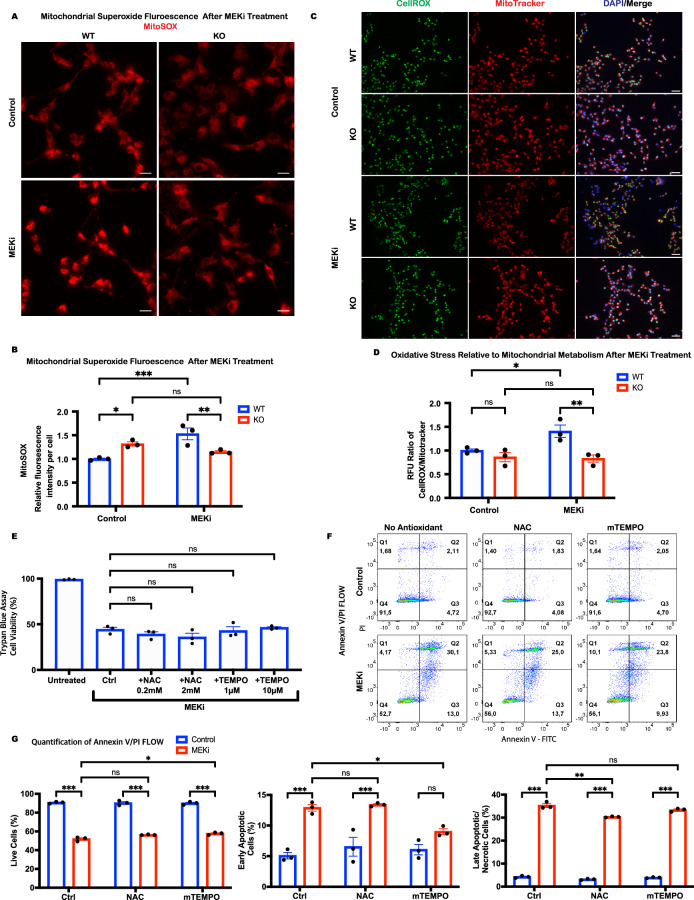
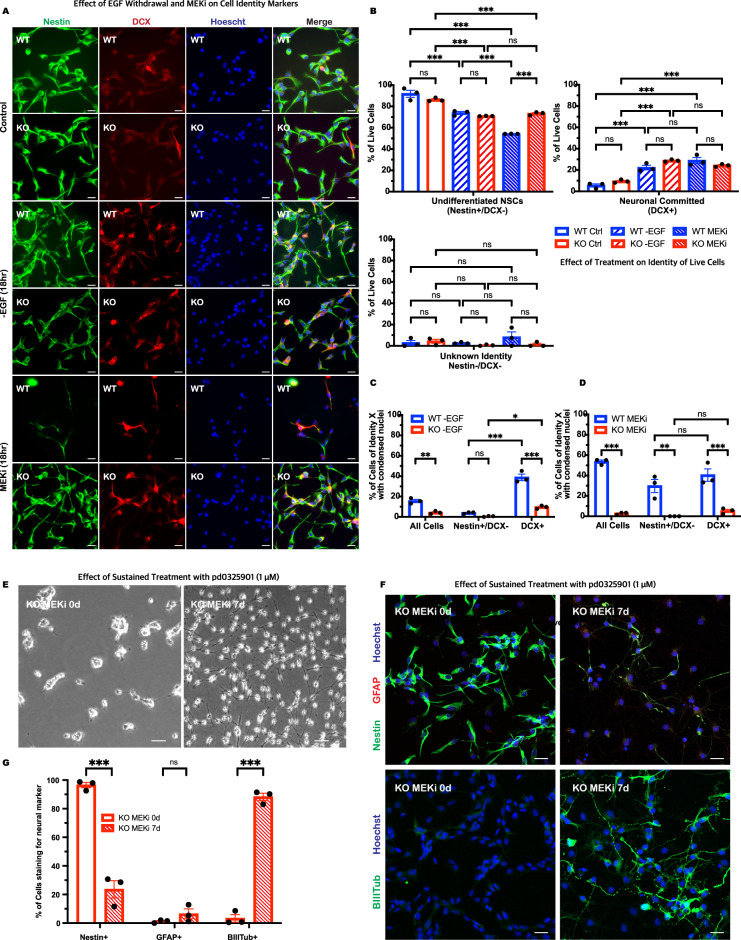
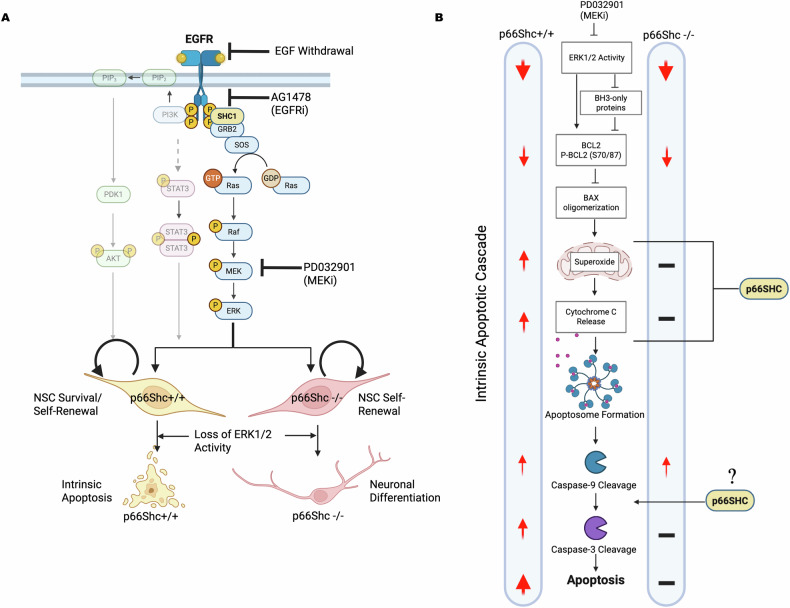
Growth factor signalling, through epidermal growth factor (EGF) and its receptor (EGFR), governs neural stem cell (NSC) proliferation, differentiation, and survival. The Src Homology and Collagen (SHC1) adaptor protein mediates EGFR survival-signalling in NSCs via its two shorter isoforms. However, the role of its longest isoform, p66Shc, in NSCs remains unclear. In this study, we investigated the role of p66Shc in NSC apoptosis by generating p66Shc knockout (p66KO) NSCs and assessing their responses to EGF withdrawal, EGFR inhibition, and MEK inhibition. We found that p66KO NSCs resisted apoptosis induced by EGF deprivation and EGFR-ERK pathway inhibition. In contrast, p66KO NSCs maintained their sensitivity to staurosporine, a general apoptosis inducer. Furthermore, p66KO NSCs subjected to prolonged MEK inhibition continued to differentiate into neurons, demonstrating their ability to evade apoptosis and progress through neuronal differentiation. These findings identify p66Shc as a pivotal regulator of NSC apoptosis in response to disrupted EGFR-ERK signalling. The ability of p66KO NSCs to resist apoptosis and differentiate without EGFR-ERK signalling highlights the potential of targeting p66Shc in conditions where growth factor signalling is disrupted, such as neurodegenerative diseases or brain injuries. Additionally, the role of p66Shc in modulating survival pathways may have broader implications for NSC-like cancers, where assessing p66Shc levels could provide prognostic value for the sensitivity of cancers to EGFR- or MEK-inhibition-based chemotherapies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: All methods were performed in accordance with the relevant guidelines and regulations. This study did not involve human participants, human-derived materials, or live vertebrate animals, and therefore did not require ethics approval or informed consent.
Figures
Similar articles
-
Transcription Factor EB Overexpression through Glial Fibrillary Acidic Protein Promoter Disrupts Neuronal Lamination by Dysregulating Neurogenesis during Embryonic Development.Dev Neurosci. 2025;47(1):40-54. doi: 10.1159/000538656. Epub 2024 Apr 18. Dev Neurosci. 2025. PMID: 38583418 Free PMC article.
-
Caveolin-1 inhibits the proliferation and invasion of lung adenocarcinoma via EGFR degradation.Sci Rep. 2025 Jul 1;15(1):21654. doi: 10.1038/s41598-025-05259-8. Sci Rep. 2025. PMID: 40594106 Free PMC article.
-
NEP1-40 Regulates the Development of Hippocampal Neural Stem Cells in Schizophrenic Mice.Curr Med Sci. 2025 Aug;45(4):930-943. doi: 10.1007/s11596-025-00078-4. Epub 2025 Jul 11. Curr Med Sci. 2025. PMID: 40643877
-
The Role of p66Shc in Cancer: Molecular Mechanisms and Therapeutic Implications.J Cell Mol Med. 2025 Jul;29(14):e70737. doi: 10.1111/jcmm.70737. J Cell Mol Med. 2025. PMID: 40690563 Free PMC article. Review.
-
A systematic review of p53 regulation of oxidative stress in skeletal muscle.Redox Rep. 2018 Dec;23(1):100-117. doi: 10.1080/13510002.2017.1416773. Epub 2018 Jan 3. Redox Rep. 2018. PMID: 29298131 Free PMC article.
References
-
- Yeo W, Gautier J. Early neural cell death: dying to become neurons. Dev Biol. 2004;274:233–44. - PubMed
-
- Miguel-Aliaga I, Thor S. Programmed cell death in the nervous system—a programmed cell fate?. Curr Opin Neurobiol. 2009;19:127–33. - PubMed
-
- Roth KA, D’Sa C. Apoptosis and brain development. Ment Retard Dev Disabil Res Rev. 2001;7:261–6. - PubMed
MeSH terms
Substances
Grants and funding
- RGPIN/04027-2019/Gouvernement du Canada | Natural Sciences and Engineering Research Council of Canada (Conseil de Recherches en Sciences Naturelles et en Génie du Canada)
- RGPIN/06893-2019/Gouvernement du Canada | Natural Sciences and Engineering Research Council of Canada (Conseil de Recherches en Sciences Naturelles et en Génie du Canada)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
