

An improved reference library and method for accurate cell-type deconvolution of bulk-tissue miRNA data
- PMID: 40593558
- PMCID: PMC12215801
- DOI: 10.1038/s41467-025-60521-x
An improved reference library and method for accurate cell-type deconvolution of bulk-tissue miRNA data
Abstract
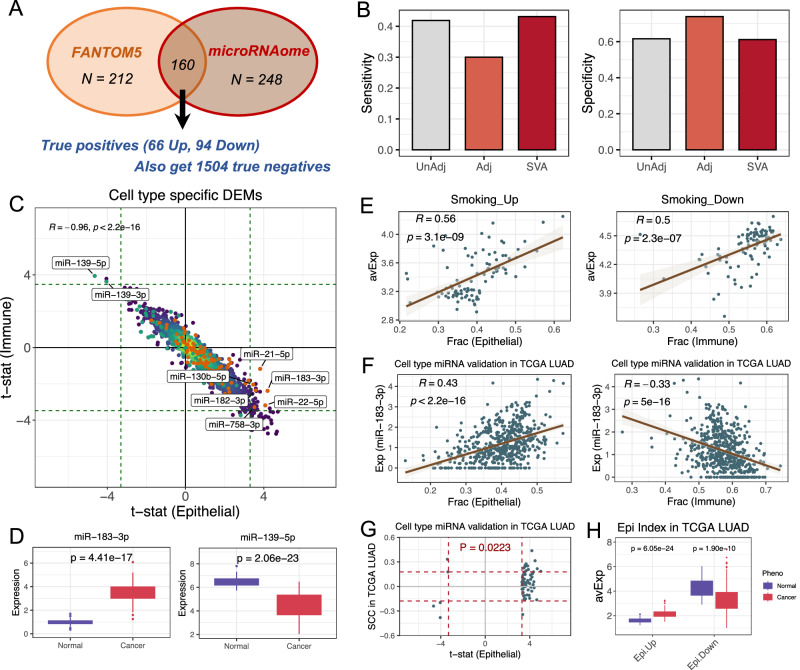
MicroRNAs (miRNAs) play key roles in development and disease, and have great biomarker potential. However, because miRNA expression is highly cell-type specific, identifying miRNA biomarkers from complex tissues is hampered by the underlying cell-type heterogeneity. Due to that current single-cell RNA-Seq protocols are lagging behind for quantification of miRNA expression, and most miRNA profiling samples do not have matched mRNA expression or DNA methylation data for cell-type deconvolution, it is an urgent need to develop computational methods for cell-type proportion estimation of bulk-tissue miRNA data. Here we present a novel miRNA expression reference library and deconvolution tool for cell-type composition estimation of complex tissues. We show that our tool is accurate and robust for deconvolution in whole blood as well as in different solid tissues. By applying this tool to a range of different biological contexts, we demonstrate its value for screening of age-associated miRNAs, for monitoring the immune landscape in infectious diseases like COVID-19, as well as for identifying cell-type-specific miRNA biomarkers for early diagnosis and prognosis of human cancers. Our work establishes a computational framework for accurate cell-type mixture deconvolution of miRNA data.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





Similar articles
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
A comprehensive investigation of identifying miRNA biomarkers and their potential role in age-related cataract by meta-analysis and bioinformatics analysis.Graefes Arch Clin Exp Ophthalmol. 2025 May;263(5):1307-1325. doi: 10.1007/s00417-024-06723-3. Epub 2025 Jan 6. Graefes Arch Clin Exp Ophthalmol. 2025. PMID: 39760860
-
Antibody tests for identification of current and past infection with SARS-CoV-2.Cochrane Database Syst Rev. 2022 Nov 17;11(11):CD013652. doi: 10.1002/14651858.CD013652.pub2. Cochrane Database Syst Rev. 2022. PMID: 36394900 Free PMC article.
-
MuSiC2: cell-type deconvolution for multi-condition bulk RNA-seq data.Brief Bioinform. 2022 Nov 19;23(6):bbac430. doi: 10.1093/bib/bbac430. Brief Bioinform. 2022. PMID: 36208175 Free PMC article.
-
Rapid, point-of-care antigen tests for diagnosis of SARS-CoV-2 infection.Cochrane Database Syst Rev. 2022 Jul 22;7(7):CD013705. doi: 10.1002/14651858.CD013705.pub3. Cochrane Database Syst Rev. 2022. PMID: 35866452 Free PMC article.
References
-
- Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell116, 281–297 (2004). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
