

Cardiomyocyte-derived USP13 protects hearts from hypertrophy via deubiquitinating and stabilizing STAT1 in male mice
- PMID: 40593642
- PMCID: PMC12217623
- DOI: 10.1038/s41467-025-61028-1
Cardiomyocyte-derived USP13 protects hearts from hypertrophy via deubiquitinating and stabilizing STAT1 in male mice
Abstract
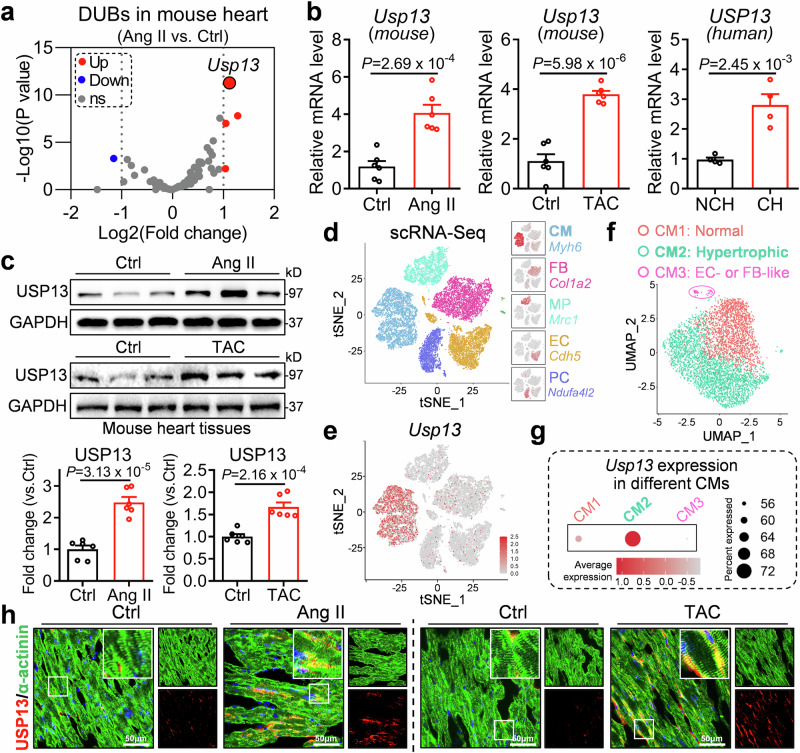
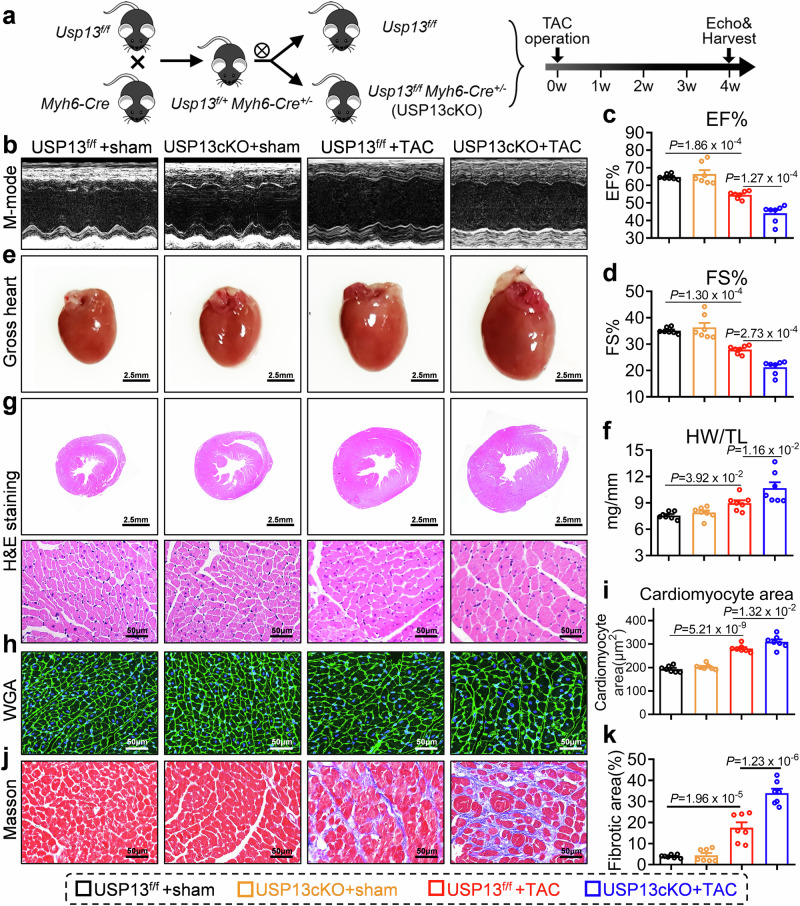
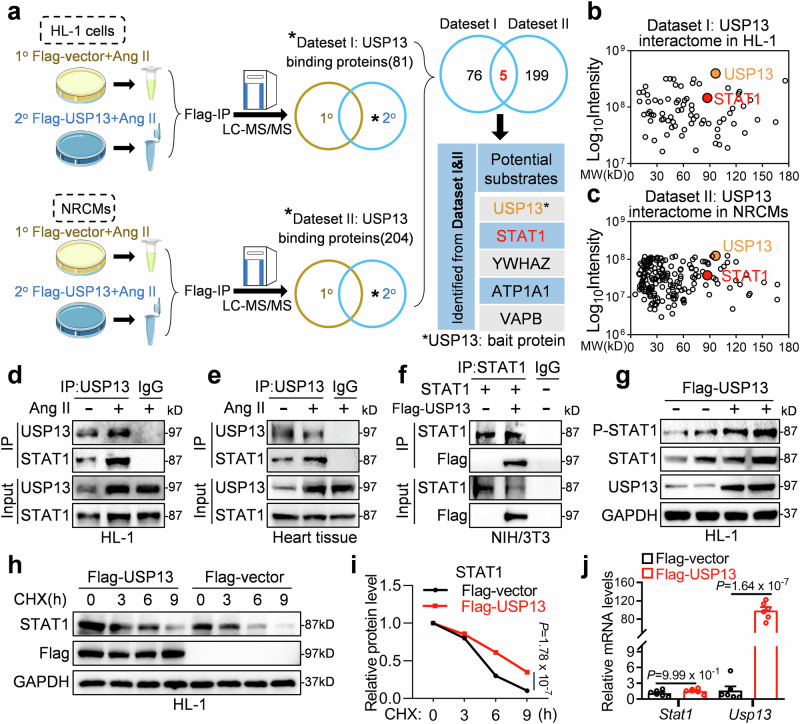
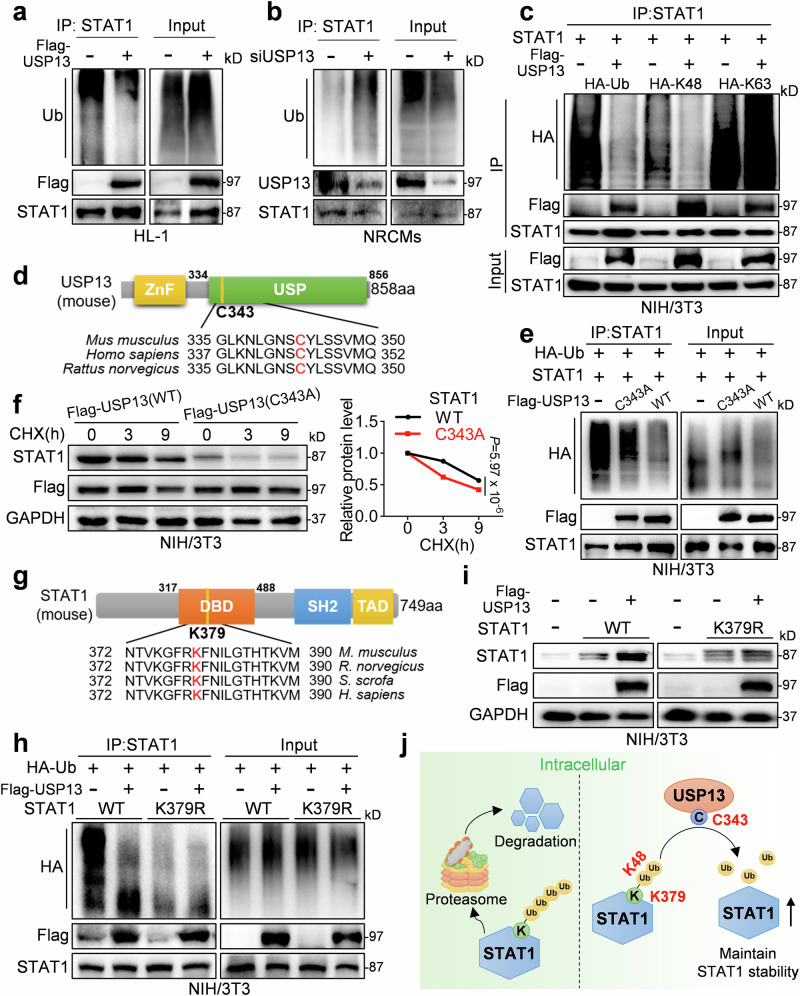
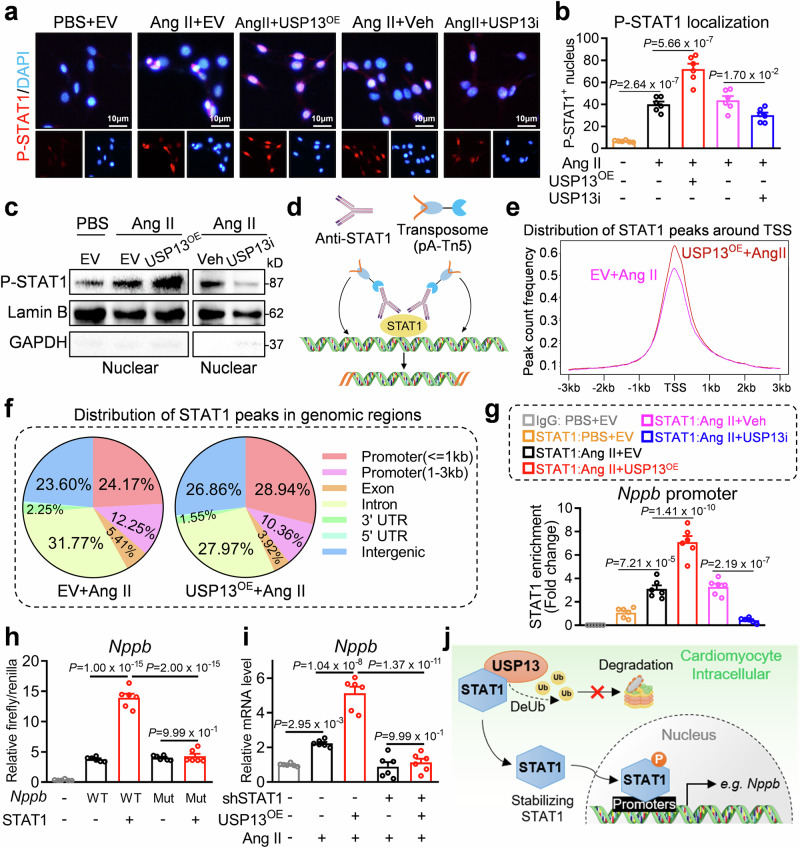
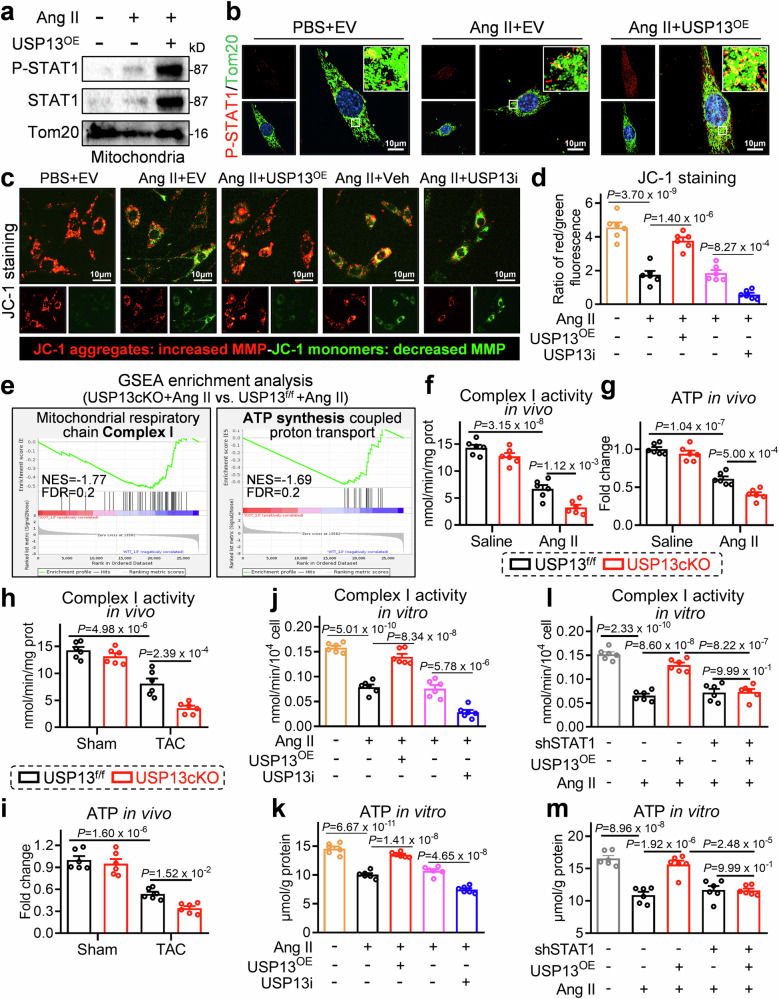
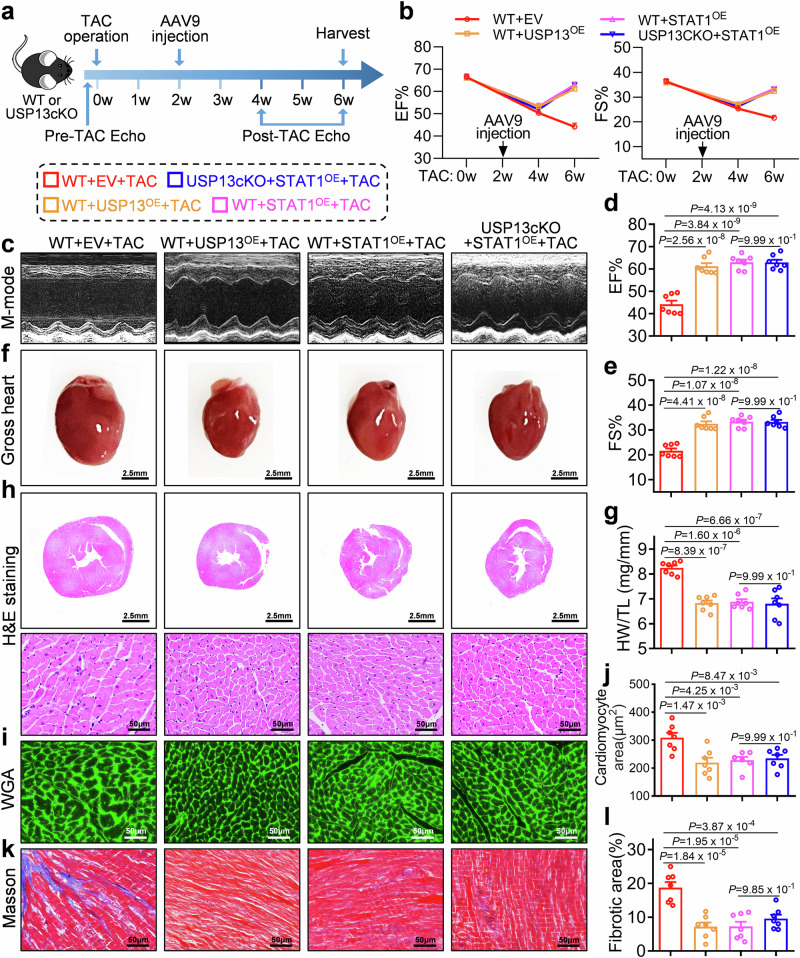
Cardiac hypertrophy leads to ventricular dysfunction and heart failure. Deubiquitinating enzymes are responsible for preserving the substrate protein stability and are essential to myocardial hypertrophy. In this study, we aimed to explore the role and regulatory mechanism of a cardiomyocyte-derived deubiquitinating enzyme, USP13, in cardiac hypertrophy. Here we show that USP13 was increased in hypertrophic myocardium and was mainly distributed in cardiomyocytes. Cardiomyocyte-specific Usp13 knockout aggravated TAC or Ang II-induced myocardial hypertrophy and dysfunction in male mice. Correspondingly, USP13 overexpression by AAV9 in hearts exerted a therapeutic impact on cardiac hypertrophy in male mice. Mechanistically, we identified STAT1 as a substrate of USP13 through interactome analysis. USP13 deubiquitinated STAT1, thereby reducing its degradation. Subsequently, USP13 promoted the STAT1-targeted Nppb gene transcription and enhanced mitochondrial function in cardiomyocytes. This study illustrated a beneficial effect of USP13 in hypertrophic cardiomyocytes and identified a cardiomyocyte-specific USP13-STAT1 axis in regulating cardiac hypertrophy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
TNIP3 protects against pathological cardiac hypertrophy by stabilizing STAT1.Cell Death Dis. 2024 Jun 26;15(6):450. doi: 10.1038/s41419-024-06805-4. Cell Death Dis. 2024. PMID: 38926347 Free PMC article.
-
Cardiomyocyte-derived YOD1 promotes pathological cardiac hypertrophy by deubiquitinating and stabilizing STAT3.Sci Adv. 2025 Jun 27;11(26):eadu8422. doi: 10.1126/sciadv.adu8422. Epub 2025 Jun 25. Sci Adv. 2025. PMID: 40561034 Free PMC article.
-
Cardiomyocyte-Enriched USP20 Ameliorates Pathological Cardiac Hypertrophy by Targeting STAT3 Deubiquitination.Adv Sci (Weinh). 2025 Jun;12(23):e2416478. doi: 10.1002/advs.202416478. Epub 2025 Apr 7. Adv Sci (Weinh). 2025. PMID: 40192103 Free PMC article.
-
Laser therapy for treating hypertrophic and keloid scars.Cochrane Database Syst Rev. 2022 Sep 26;9(9):CD011642. doi: 10.1002/14651858.CD011642.pub2. Cochrane Database Syst Rev. 2022. PMID: 36161591 Free PMC article.
-
Coenzyme Q10 for heart failure.Cochrane Database Syst Rev. 2021 Feb 3;(2)(2):CD008684. doi: 10.1002/14651858.CD008684.pub3. Cochrane Database Syst Rev. 2021. PMID: 35608922 Free PMC article.
References
-
- Nakamura, M. & Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol.15, 387–407 (2018). - PubMed
-
- McKinsey, T. A. & Kass, D. A. Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nat. Rev. Drug Discov.6, 617–635 (2007). - PubMed
-
- Popovic, D., Vucic, D. & Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med.20, 1242–1253 (2014). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
