

Mapping the nuclear landscape with multiplexed super-resolution fluorescence microscopy
- PMID: 40593784
- PMCID: PMC12215941
- DOI: 10.1038/s41467-025-61358-0
Mapping the nuclear landscape with multiplexed super-resolution fluorescence microscopy
Abstract
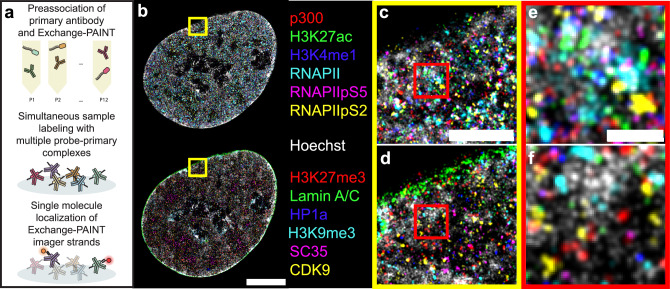
The nucleus coordinates many different processes. Visualizing how these are spatially organized requires imaging protein complexes, epigenetic marks, and DNA across scales from single molecules to the whole nucleus. To accomplish this, we develop a multiplexed imaging protocol to localize 13 different nuclear targets with nanometer precision. Within single cells, we show that nuclear specification into active and repressive states exists along a spectrum of length scales, emerging below one micron and becoming strengthened at the nanoscale with unique organizational principles in both heterochromatin and euchromatin. HP1α was positively correlated with DNA at the microscale but uncorrelated at the nanoscale. RNA Polymerase II, p300, and CDK9 were positively correlated at the microscale but became partitioned below 300 nm. Perturbing histone acetylation or transcription disrupted nanoscale organization but had less effect at the microscale. We envision that our imaging and analysis pipeline will be useful to reveal the organizational principles not only of the cell nucleus but also other cellular compartments.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures







Update of
-
Mapping the nuclear landscape with multiplexed super-resolution fluorescence microscopy.bioRxiv [Preprint]. 2024 Jul 29:2024.07.27.605159. doi: 10.1101/2024.07.27.605159. bioRxiv. 2024. Update in: Nat Commun. 2025 Jul 1;16(1):6042. doi: 10.1038/s41467-025-61358-0. PMID: 39211261 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
