

Triglycerides are an important fuel reserve for synapse function in the brain
- PMID: 40595405
- PMCID: PMC12286841
- DOI: 10.1038/s42255-025-01321-x
Triglycerides are an important fuel reserve for synapse function in the brain
Abstract
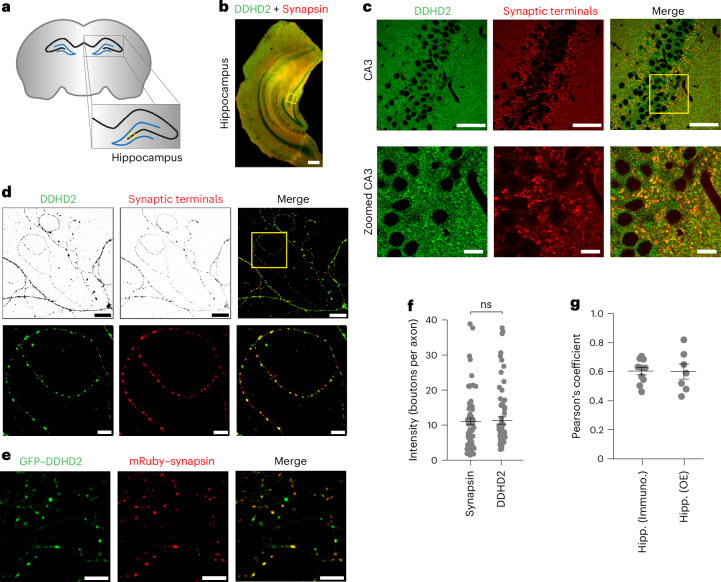
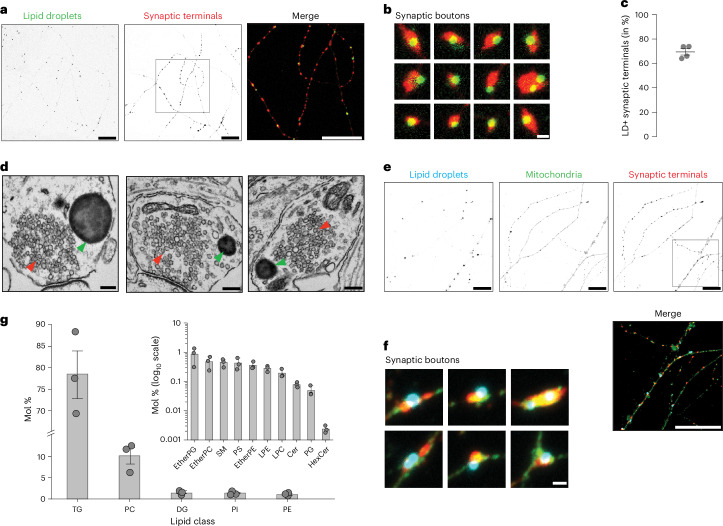
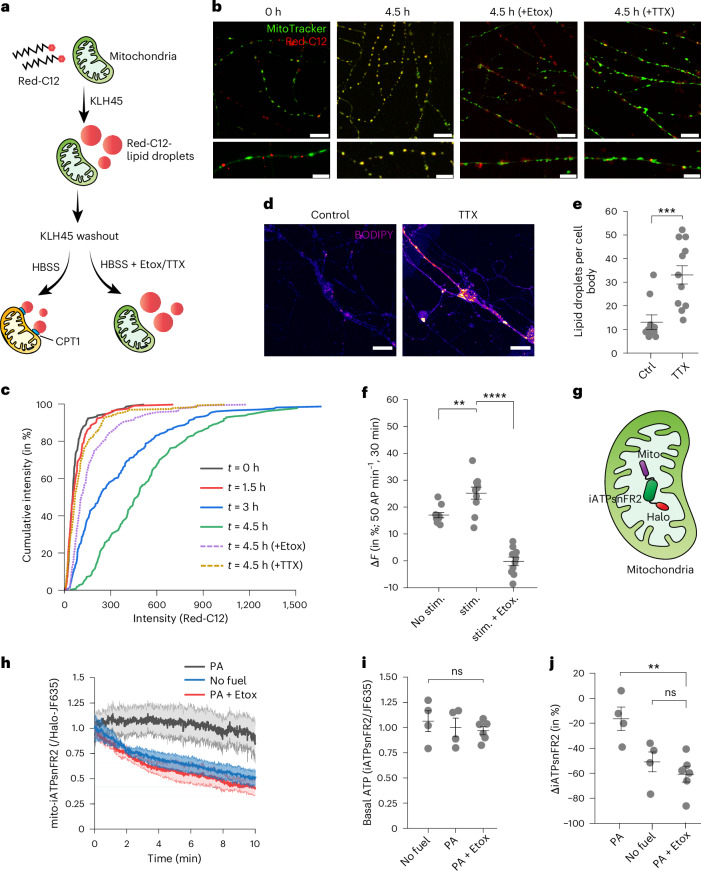
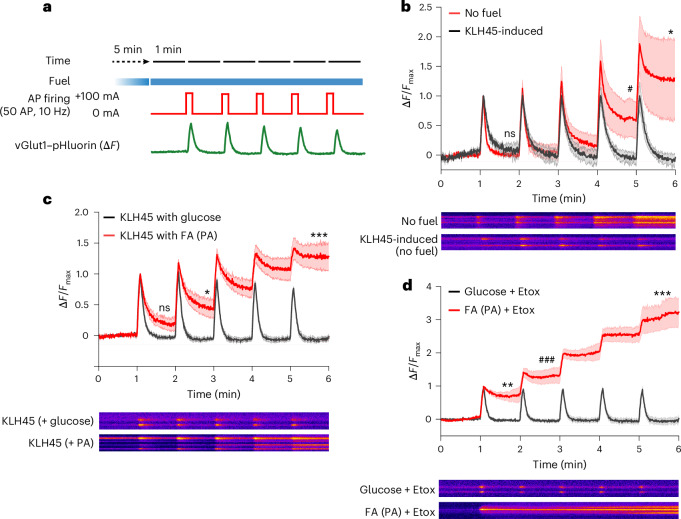
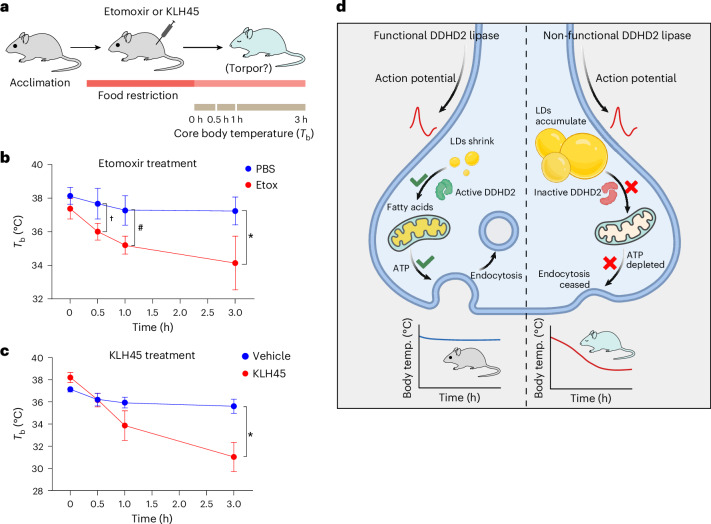
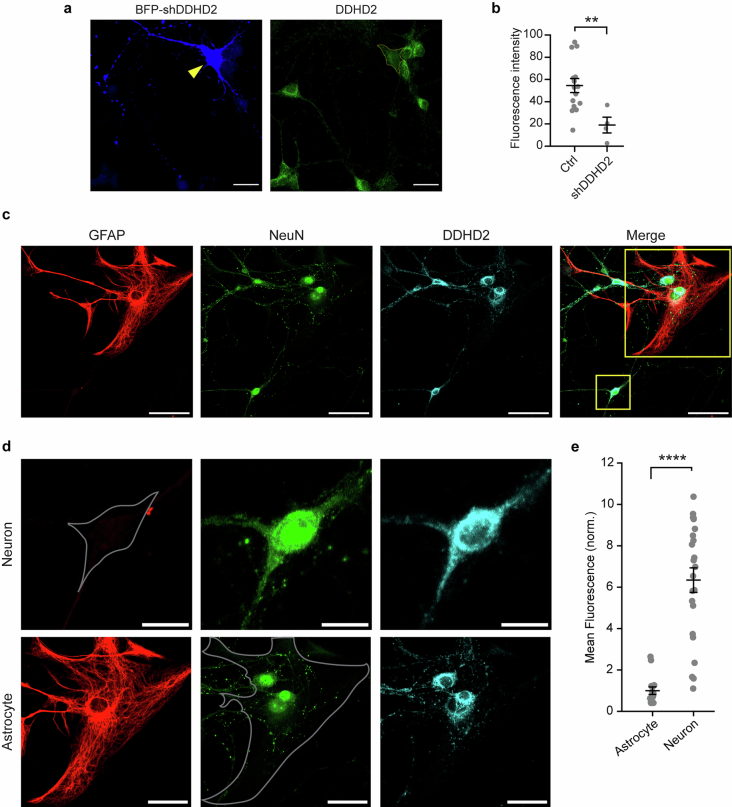
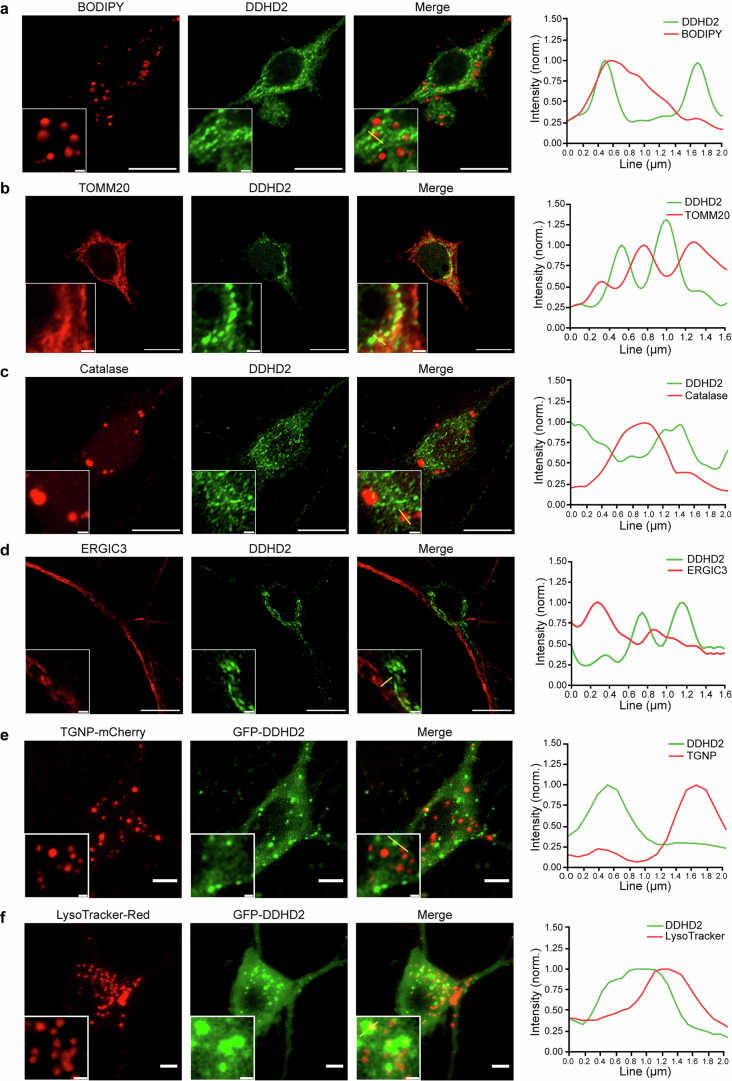
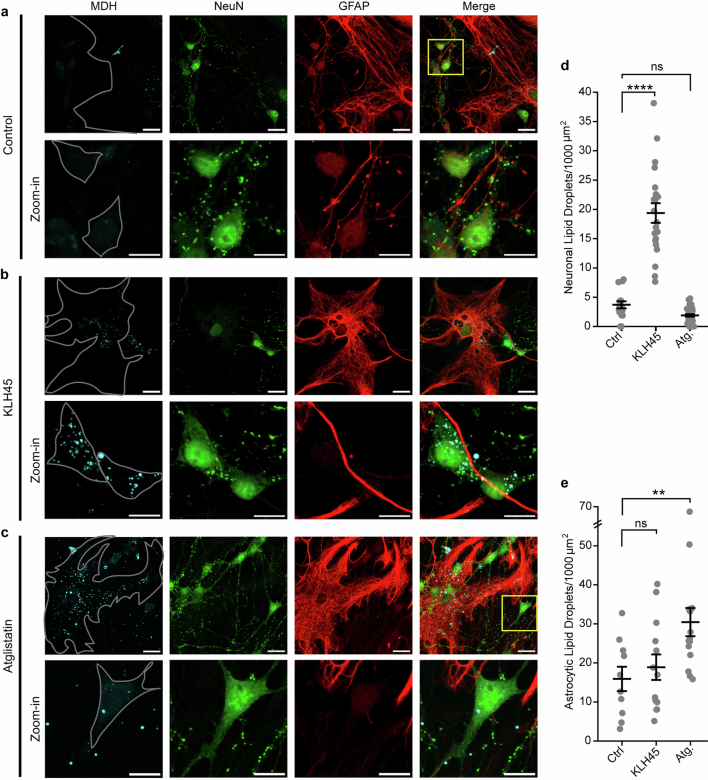
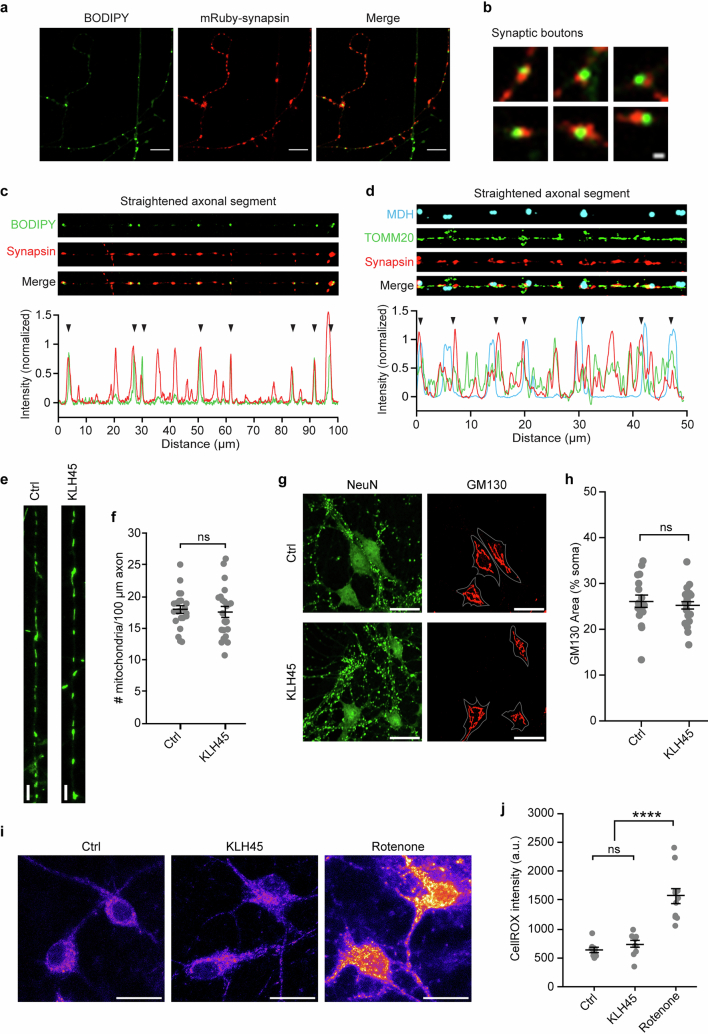
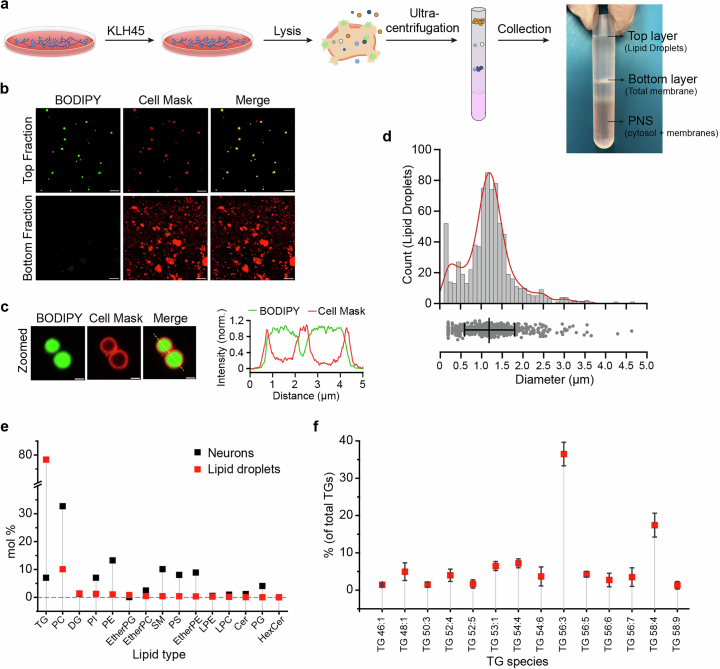
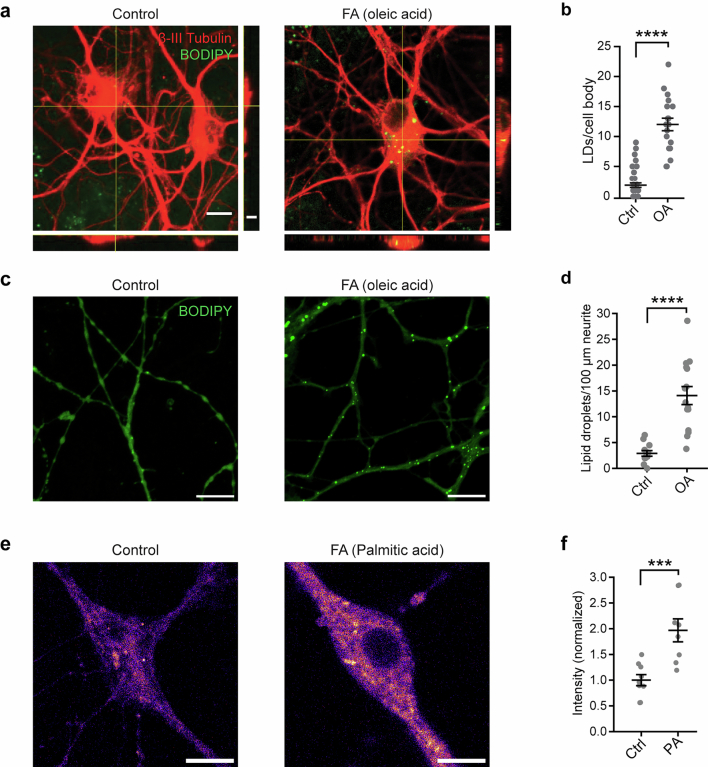
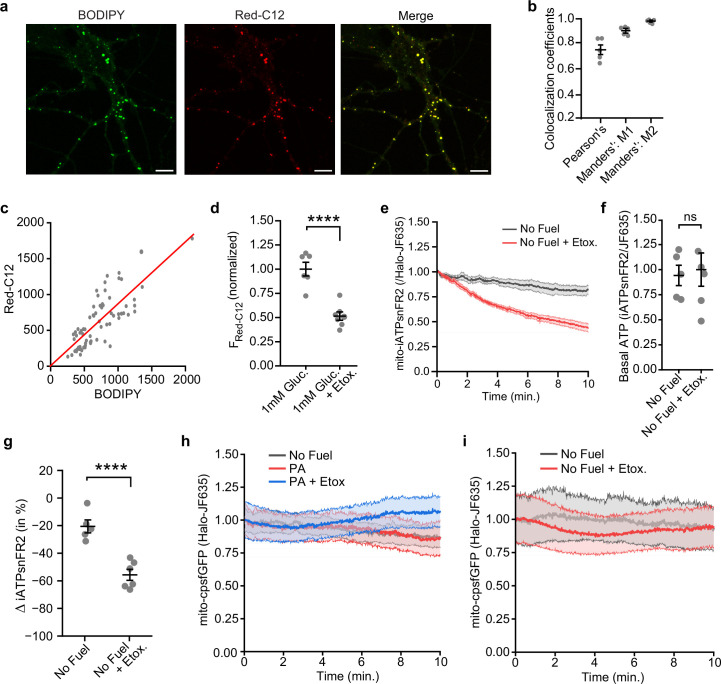
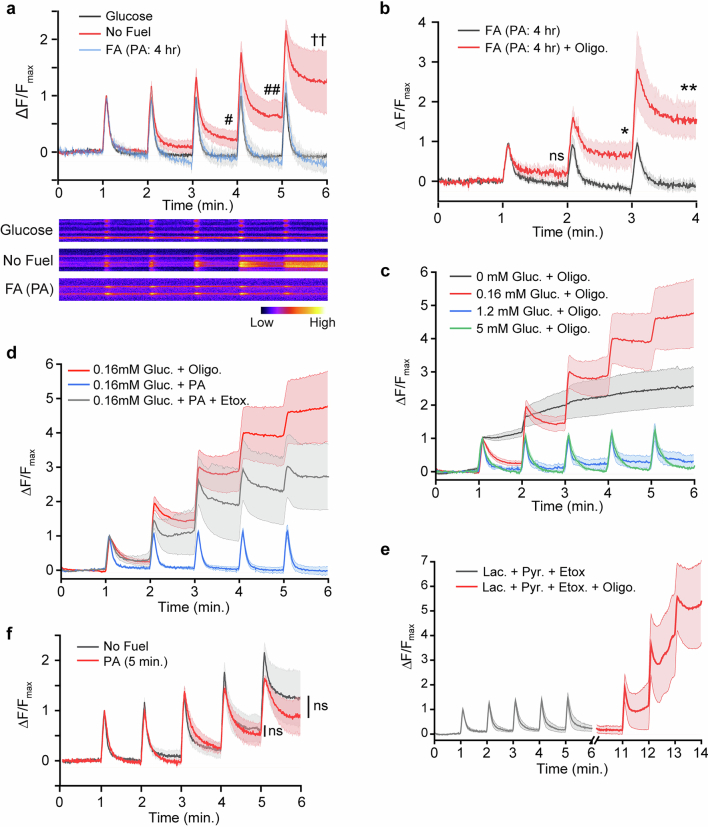
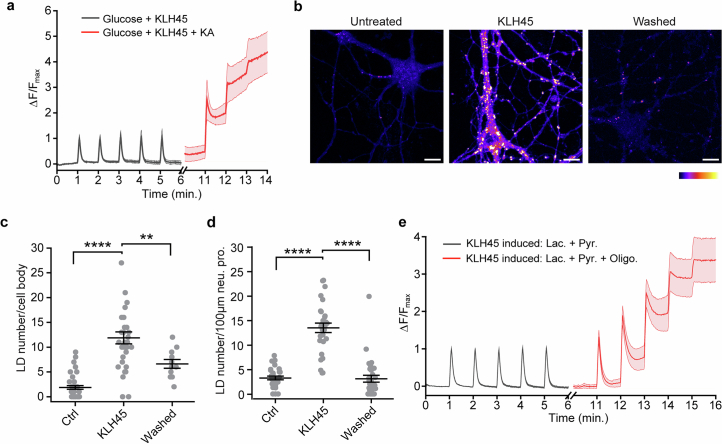
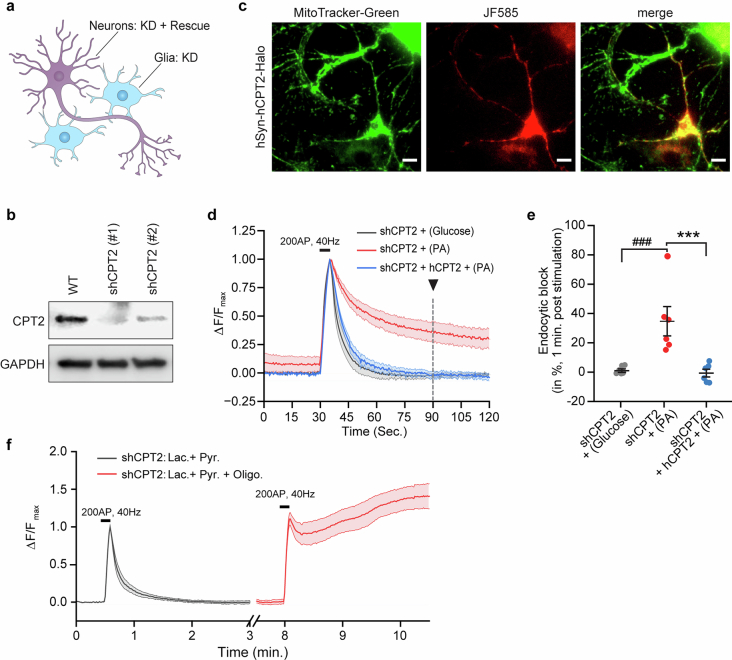
Proper fuelling of the brain is critical to sustain cognitive function, but the role of fatty acid (FA) combustion in this process has been elusive. Here we show that acute block of a neuron-specific triglyceride lipase, DDHD2 (a genetic driver of complex hereditary spastic paraplegia), or of the mitochondrial lipid transporter CPT1 leads to rapid onset of torpor in adult male mice. These data indicate that in vivo neurons are probably constantly fluxing FAs derived from lipid droplets (LDs) through β-oxidation to support neuronal bioenergetics. We show that in dissociated neurons, electrical silencing or blocking of DDHD2 leads to accumulation of neuronal LDs, including at nerve terminals, and that FAs derived from axonal LDs enter mitochondria in an activity-dependent fashion to drive local mitochondrial ATP production. These data demonstrate that nerve terminals can make use of LDs during electrical activity to provide metabolic support and probably have a critical role in supporting neuron function in vivo.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
