

Hydroxychloroquine sulfate for IgA nephropathy: mechanisms and therapeutic potential in improving proteinuria and alleviating disease progression - a literature review
- PMID: 40597811
- PMCID: PMC12210545
- DOI: 10.1186/s12882-025-04262-5
Hydroxychloroquine sulfate for IgA nephropathy: mechanisms and therapeutic potential in improving proteinuria and alleviating disease progression - a literature review
Abstract
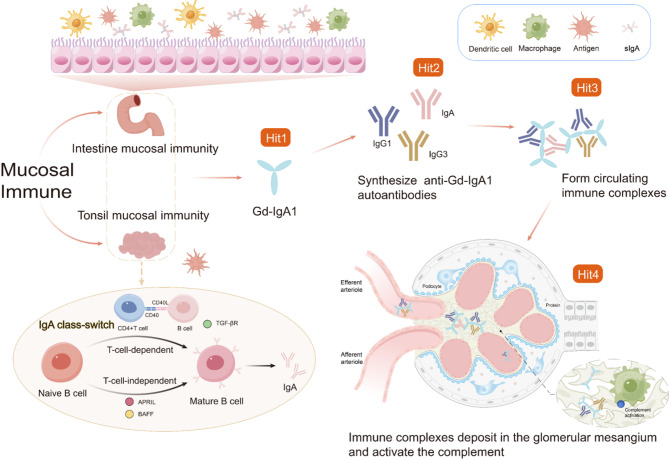
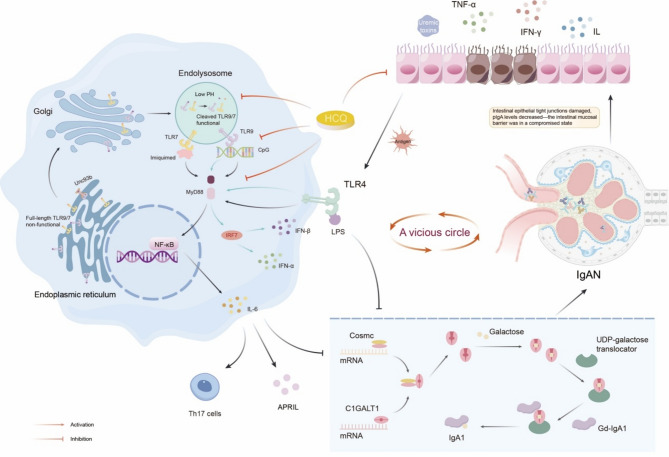
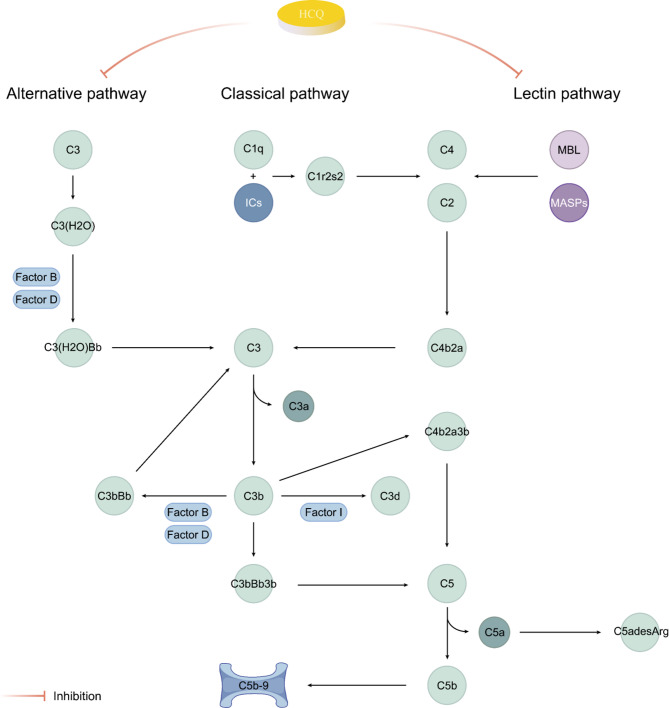
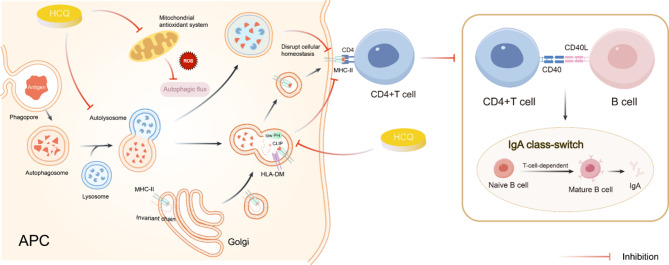
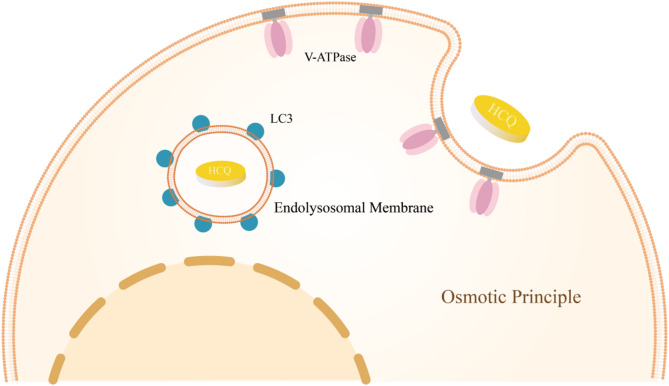
IgA nephropathy (IgAN), the most common form of glomerulonephritis worldwide, often progresses to chronic kidney failure within 10 to 15 years. Despite its clinical importance, effective disease-modifying therapies for IgAN remain limited. Proteinuria is well recognized as both a prognostic biomarker and a modifiable therapeutic target in IgAN. Several randomized controlled trials conducted among Chinese patients with IgAN have demonstrated the efficacy of hydroxychloroquine (HCQ) in reducing proteinuria. The Kidney Disease: Improving Global Outcomes (KDIGO) guidelines also suggest that HCQ may exert potential therapeutic effects in IgAN. However, the molecular mechanisms underlying the renoprotective effects of HCQ remain incompletely understood. This review synthesises current evidence on HCQ's therapeutic mechanisms in IgAN, highlighting its multifaceted roles in: (1) suppressing pathogenic galactose-deficient IgA1 synthesis through modulation of mucosal immunity, Toll-like receptor (TLR) signaling, IL-6 pathways, and complement activation; (2) inhibiting autophagy-mediated antigen presentation via major histocompatibility complex class II (MHC-II) molecules; (3) modulating non-canonical autophagy pathways to attenuate human mesangial cells (HMCs) proliferation and protect podocytes; and (4) demonstrating antithrombotic effects. Collectively, HCQ demonstrates multifaceted mechanisms for proteinuria reduction in IgAN while maintaining a favorable safety profile.
Not applicable.
Keywords: Galactose-deficient IgA1; Hydroxychloroquine; IgA nephropathy; Mechanism; Proteinuria.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics, consent to participate, and consent to publish: Not applicable. Literature search strategy: We have augmented our database searches with Web of Science and PubMed for “IgAN,” “Proteinuria,” “HCQ,” “Gd-IgA1,” “Autophagy,” and related keywords, with a focus on literature published up to 2025. This approach has enabled us to comprehensively track down references to key literature. The literature was initially assessed by title and abstract, followed by a thorough review of the full text to ascertain its relevance. Ultimately, a decision regarding inclusion was made based on the study’s quality, methodological rigor, and its pertinence to the subject matter. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Exploring the mechanism of proteinuria reduction by hydroxychloroquine in IgA nephropathy using network pharmacology and molecular mocking.Sci Rep. 2025 Jul 1;15(1):20836. doi: 10.1038/s41598-025-97950-z. Sci Rep. 2025. PMID: 40593236 Free PMC article.
-
Different doses of hydroxychloroquine regulate the structure of intestinal flora and glycosyltransferase activity in rats with IgA nephropathy.Immunobiology. 2025 May;230(3):152891. doi: 10.1016/j.imbio.2025.152891. Epub 2025 Mar 12. Immunobiology. 2025. PMID: 40112730
-
Dynamics of chromatin accessibility governing Gd-IgA1 synthesis in B cells associated with IgA nephropathy.Exp Mol Med. 2025 Jul;57(7):1593-1606. doi: 10.1038/s12276-025-01505-1. Epub 2025 Jul 23. Exp Mol Med. 2025. PMID: 40702134 Free PMC article.
-
Efficacy and safety of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for IgA nephropathy in children.Pediatr Nephrol. 2022 Mar;37(3):499-508. doi: 10.1007/s00467-021-05316-0. Epub 2021 Oct 22. Pediatr Nephrol. 2022. PMID: 34686915
-
Non-immunosuppressive treatment for IgA nephropathy.Cochrane Database Syst Rev. 2011 Mar 16;(3):CD003962. doi: 10.1002/14651858.CD003962.pub2. Cochrane Database Syst Rev. 2011. Update in: Cochrane Database Syst Rev. 2024 Feb 1;2:CD003962. doi: 10.1002/14651858.CD003962.pub3. PMID: 21412884 Updated.
References
-
- Stamellou E, Seikrit C, Tang SCW, et al. IgA nephropathy. Nat Rev Dis Primers. 2023;9(1):67. 10.1038/s41572-023-00476-9. - PubMed
-
- de Zhou F, Zhao Mhui, Zou W, zhong, Liu G, Wang H. The changing spectrum of primary glomerular diseases within 15 years: a survey of 3331 patients in a single Chinese centre. Nephrol Dial Transpl. 2009;24(3):870–6. 10.1093/ndt/gfn554. - PubMed
-
- Li LS, Liu ZH. Epidemiologic data of renal diseases from a single unit in china: analysis based on 13,519 renal biopsies. Kidney Int. 2004;66(3):920–3. 10.1111/j.1523-1755.2004.00837.x. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 81573791/National Natural Science Foundation of China
- 81973683/National Natural Science Foundation of China,China
- 82104666/Youth Fund of the National Natural Science Foundation of China
- CI2023C051YLL/Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences
- ZZ15-YQ-025/the Excellent Young Scientific and Technological Talents (Innovation) Training Project of China Academy of Chinese Medical Sciences
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
