

The role and mechanism of TSC in kidney diseases: a literature review
- PMID: 40597820
- PMCID: PMC12210502
- DOI: 10.1186/s12882-025-04260-7
The role and mechanism of TSC in kidney diseases: a literature review
Abstract
Background: Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by multisystem involvement, primarily caused by loss-of-function mutations in the TSC1 or TSC2 genes. TSC is a key integrator of metabolic signaling and cellular stress and has become an important regulator in several kidney diseases. TSC1 and TSC2 can be used not only as genetic markers for disease diagnosis, but also as potential immunotherapeutic targets for kidney disease. Recent studies on the pathogenesis of TSC may provide guidance for developing new treatment strategies for kidney diseases.
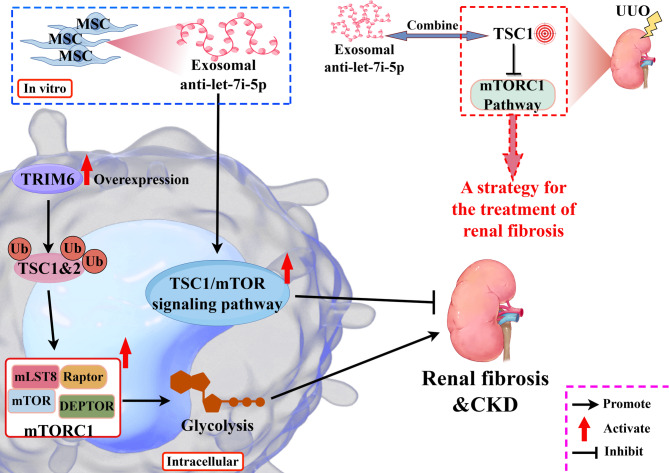
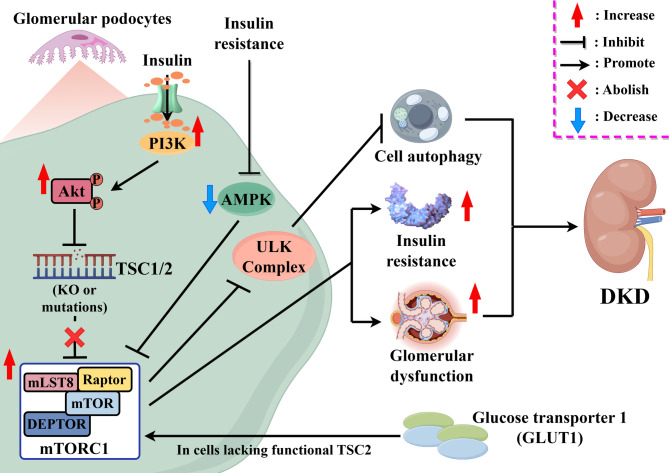
Key messages: Therefore, we systematically reviewed the molecular biology of TSC and their signaling pathway, regulation of cell metabolism, and immune response in acute renal injury, chronic kidney disease, diabetic kidney disease, renal cysts, benign and malignant intrarenal tumors, and renal angiomyolipomas. We also summarize the efficacy and adverse effects of mTOR inhibitors in the treatment of TSC-related kidney diseases.
Keywords: Immunotherapeutic; Kidney diseases; Tuberous sclerosis complex; mTOR inhibitors.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: None required for reviews. Informed consent to participate: It does not apply to reviews. Competing interests: The authors declare no competing interests.
Figures






References
-
- Eckardt K-U, Coresh J, Devuyst O, Johnson RJ, Köttgen A, Levey AS, et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013;382:158–69. 10.1016/s0140-6736(13)60439-0. - PubMed
-
- Dare AJ, Fu SH, Patra J, Rodriguez PS, Thakur JS, Jha P. Renal failure deaths and their risk factors in India 2001–13: nationally representative estimates from the million death study. Lancet Global Health. 2017;5:e89–95. 10.1016/s2214-109x(16)30308-4. - PubMed
-
- Kurts C, Meyer-Schwesinger C. Protecting the kidney against autoimmunity and inflammation. Nat Rev Nephrol. 2019;15:66–8. 10.1038/s41581-018-0095-2. - PubMed
-
- Tang PM-K, Nikolic-Paterson DJ, Lan H-Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15:144–58. 10.1038/s41581-019-0110-2. - PubMed
-
- Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Reviews Disease Primers. 2016;2:16035. 10.1038/nrdp.2016.35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
