

Comparative analysis of statistical and deep learning-based multi-omics integration for breast cancer subtype classification
- PMID: 40598554
- PMCID: PMC12210783
- DOI: 10.1186/s12967-025-06662-5
Comparative analysis of statistical and deep learning-based multi-omics integration for breast cancer subtype classification
Abstract
Background: Breast cancer (BC) is a critical cause of cancer-related death globally. The heterogeneity of BC subtypes poses challenges in understanding molecular mechanisms, early diagnosis, and disease management. Recent studies suggest that integrating multi-omics layers can significantly enhance BC subtype identification. However, evaluating different multi-omics integration methods for BC subtyping remains ambiguous.
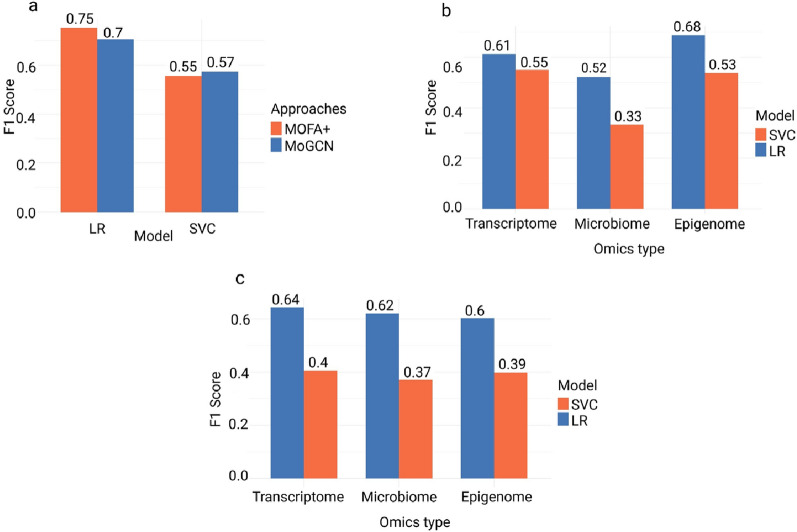
Methods: In this study, we conducted a multi-omics integration analysis on 960 BC patient samples, incorporating three omics layers: Host transcriptomics, epigenomics, and shotgun microbiome. We compared two integration approaches the statistical-based approach (MOFA+) and a deep learning-based approach (MOGCN) for this integration. We evaluated both methods using complementary evaluation criteria. First, we assessed the ability of selected features to discriminate between BC subtypes using both linear and nonlinear classification models. Second, we analyzed the biological relevance of the selected features to key BC pathways, focusing on transcriptomics-driven insights.
Results: Our results showed that MOFA+ outperformed MOGCN in feature selection, achieving the highest F1 score (0.75) in the nonlinear classification model, with MOFA+ also identifying 121 relevant pathways compared to 100 from MOGCN. Notably, one of the key pathways Fc gamma R-mediated phagocytosis and the SNARE pathway was implicated, offering insights into immune responses and tumor progression.
Conclusion: These findings suggest that MOFA+ is a more effective unsupervised tool for feature selection in BC subtyping. Our study underscores the potential of multi-omics integration to improve BC subtype prediction and provides critical insights for advancing personalized medicine in BC.
Keywords: Breast cancer; F1 score; Fc gamma R-mediated phagocytosis; MOFA+; MoGCN; Multi-omics integration; Network analysis; Personalized Medicine; SNARE pathway.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Ethical approval and consent to participate were waived since we used only publicly available data and materials in this study. Consent for publication: No consent. Competing interests: The authors declare that they have no competing interests.
Figures




References
-
- Aguilar DL, et al. Towards an interpretable autoencoder: a decision-tree-based autoencoder and its application in anomaly detection. IEEE Trans Dependable Secure Comput. 2023;20(2):1048–59. 10.1109/TDSC.2022.3148331. - DOI
-
- Argelaguet R et al. Multi-Omics factor analysis disentangles heterogeneity in blood cancer. BioRxiv. 2017. p. 217554.
-
- Bascol K et al. Unsupervised interpretable pattern discovery in time series using autoencoders. 2016. p. 427–38. 10.1007/978-3-319-49055-7_38.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
