

The genetic spectrum features of 2261 Chinese children with epilepsy and intellectual disability
- PMID: 40598568
- PMCID: PMC12219049
- DOI: 10.1186/s12916-025-04220-w
The genetic spectrum features of 2261 Chinese children with epilepsy and intellectual disability
Abstract
Background: Epilepsy (EP) and intellectual disability (ID) are two highly correlated diseases that seriously impact neurodevelopment in children. Precision diagnosis of EP and ID remains challenging due to their clinical and genetic heterogeneity, necessitating a profound understanding of disease characteristics.
Methods: We provide a clinical and genetic landscape of 2261 Chinese patients performed chromosome microarray analysis (CMA) or next-generation sequencing to uncover causal copy number variants (CNVs) or single-nucleotide variants (SNVs). Patients were stratified into three groups: EP (374 cases), ID (863 cases), and EP + ID (1024 cases).
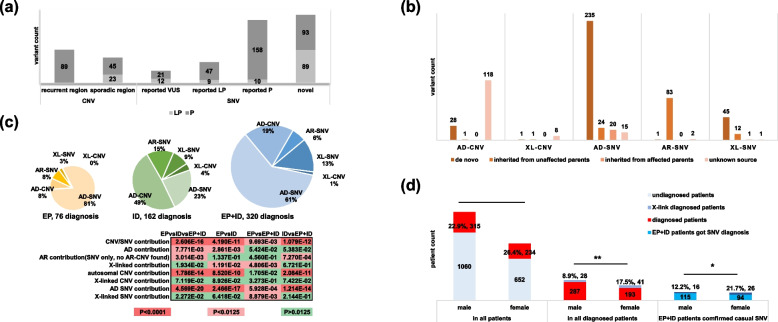
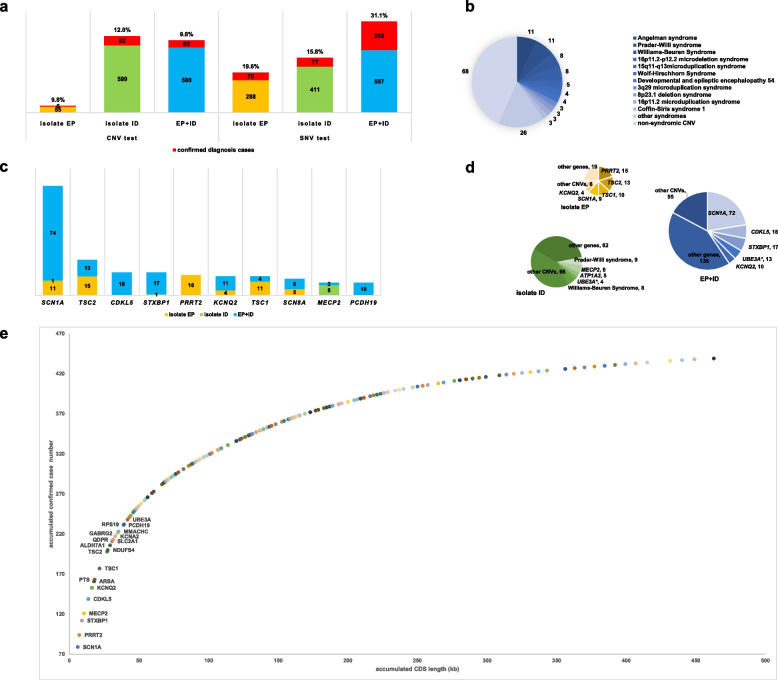
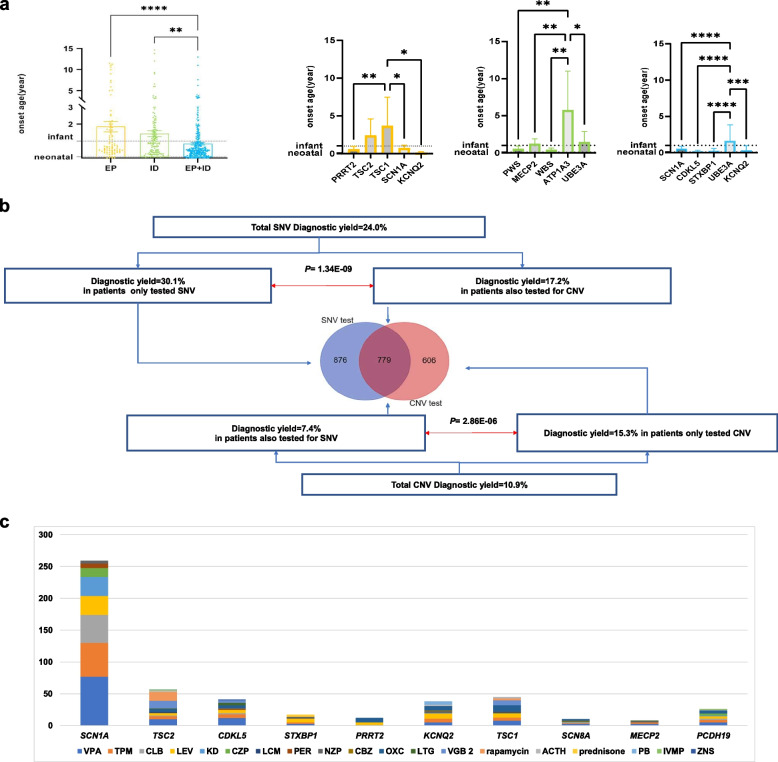
Results: We reported a 24.3% diagnostic yield from 496 causal CNVs and SNVs, including 182 novel variants, in which updated 33 previously reported VUS. Significant intergroup differences emerged: EP patients were predominantly caused by autosomal dominant SNVs, showing the highest rates of incomplete penetrance and family history. ID patients were more likely caused by CNVs and autosomal recessive SNVs, with the highest genetic heterogeneity. EP + ID patients displayed the earliest onset ages and highest diagnostic yields. We prioritized genes by diagnostic efficiency and revealed that X-linked SNVs disproportionately affected females, particularly in the EP + ID group, under current diagnostic paradigms. This real-world dataset informs genetic counseling, testing strategies, precision therapies, and long-term management for EP/ID.
Conclusions: The clinical and genetic profiles from this study provided a reliable baseline reference for diagnosing EP and ID.
Keywords: Clinical assessment; Epilepsy; Intellectual disability; Molecular diagnosis; Sex difference.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study was locally approved by the institutional review board of Xiangya Hospital of Central South University, China (201605585). All patients in this study signed the informed consent. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
-
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2003;60:367–80. - DOI - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
