

Epigenetic modifications and their roles in pediatric brain tumor formation: emerging insights from chromatin dysregulation
- PMID: 40599851
- PMCID: PMC12209359
- DOI: 10.3389/fonc.2025.1569548
Epigenetic modifications and their roles in pediatric brain tumor formation: emerging insights from chromatin dysregulation
Abstract
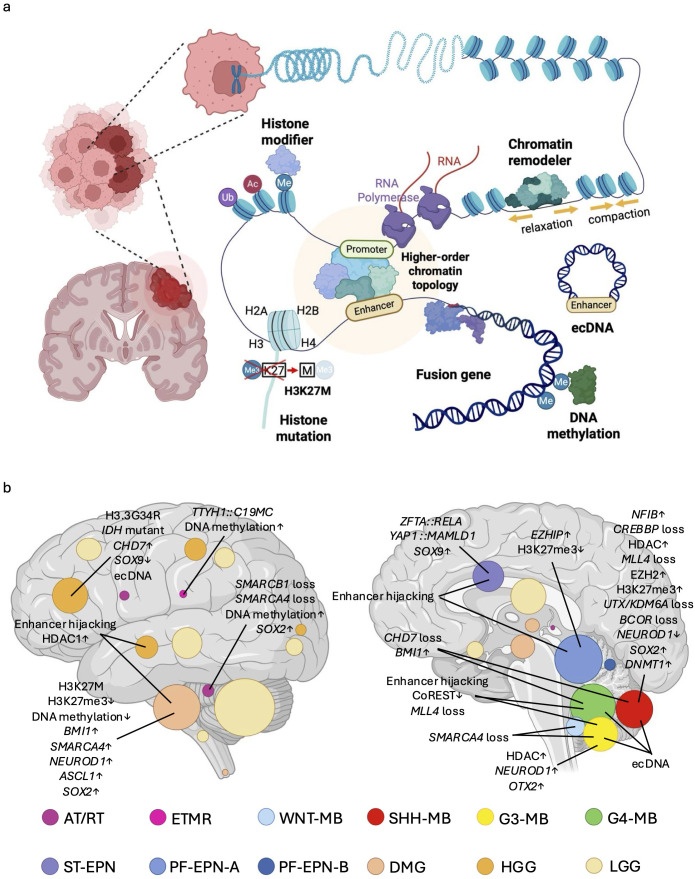
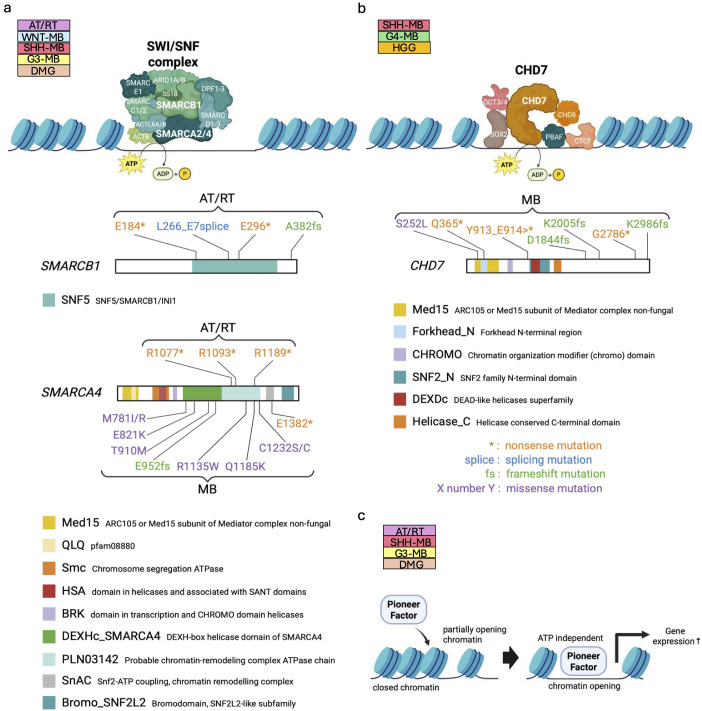
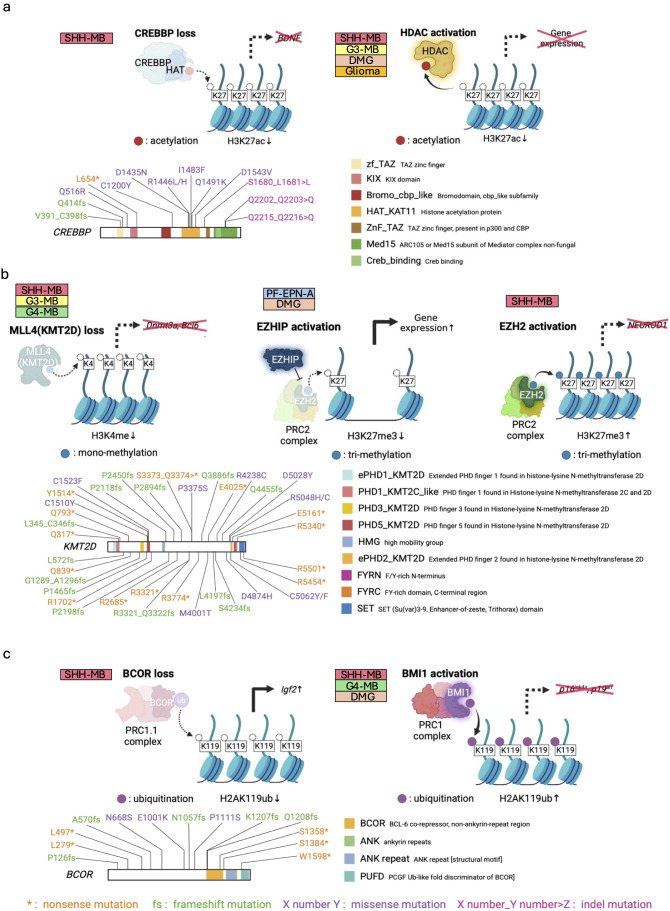
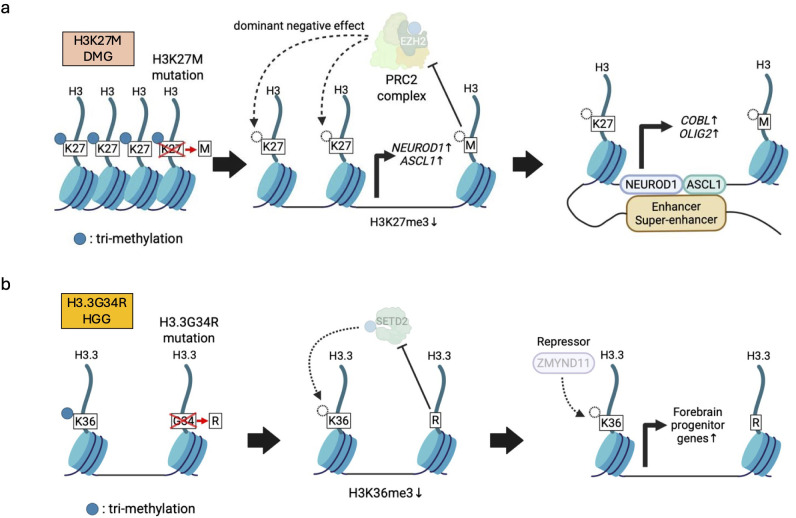
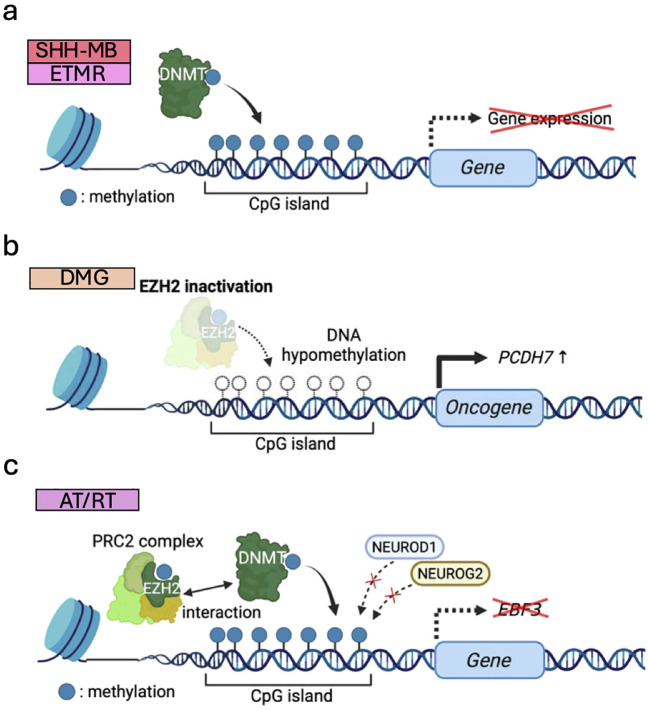
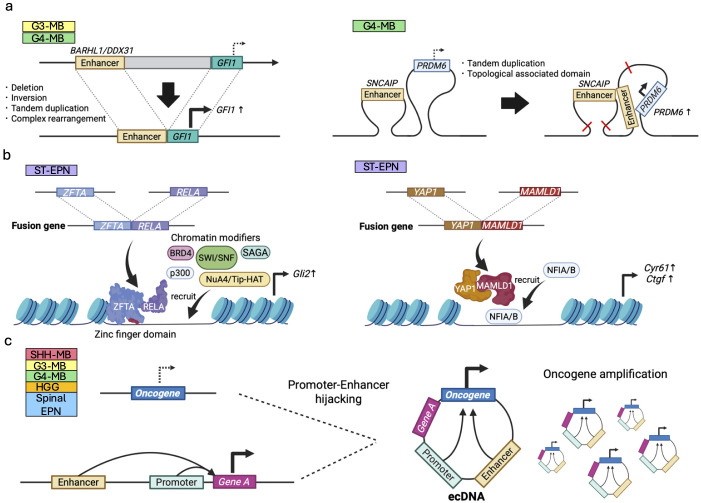
Pediatric brain tumors, the most devastating cancers affecting children, are believed to originate from neural stem/progenitor cells in developing brain. In precise timing and specific regions during the brain development, chromatin deregulation plays crucial roles in redirecting normal neuronal differentiation pathways toward tumorigenesis. Indeed, epigenomic abnormalities are thought to be more important for brain tumor formation especially in children than adults, as pediatric brain tumors generally exhibit fewer genetic mutations compared to adult brain tumors. Given the small number of mutations, targeting such limited alterations involved in cancer epigenomes is expected to be more effective in pediatric brain tumors. The mechanisms of cancer epigenomes include mutation or dysregulation of chromatin remodelers, histone modifiers, histones themselves, and DNA methylation enzymes. Furthermore, genomic rearrangements and/or higher-order chromatin topology also contribute to these epigenomic mechanisms. These mechanisms are commonly observed in various types of pediatric brain tumors. However, alterations in chromatin regulatory factors differ across tumor types, reflecting the unique epigenetic landscapes shaped by their tumor origins. Accordingly, clarifying their functional similarities and differences across tumor types could offer valuable insights for finding new therapeutic strategies. Thus, this review article focuses on elucidating how pediatric brain tumors arise from epigenomic deregulation and what epigenetic molecules or mechanisms could serve as therapeutic targets.
Keywords: DNA methylation; chromatin remodeler; epigenetics; extrachromosomal DNA (ecDNA); fusion oncogene; genomic rearrangement; histone modification; pediatric brain tumor.
Copyright © 2025 Kawata, Chapman, Narumi and Kawauchi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
[Epigenetics' implication in autism spectrum disorders: A review].Encephale. 2017 Aug;43(4):374-381. doi: 10.1016/j.encep.2016.07.007. Epub 2016 Sep 28. Encephale. 2017. PMID: 27692350 French.
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Survivor, family and professional experiences of psychosocial interventions for sexual abuse and violence: a qualitative evidence synthesis.Cochrane Database Syst Rev. 2022 Oct 4;10(10):CD013648. doi: 10.1002/14651858.CD013648.pub2. Cochrane Database Syst Rev. 2022. PMID: 36194890 Free PMC article.
-
The use of Open Dialogue in Trauma Informed Care services for mental health consumers and their family networks: A scoping review.J Psychiatr Ment Health Nurs. 2024 Aug;31(4):681-698. doi: 10.1111/jpm.13023. Epub 2024 Jan 17. J Psychiatr Ment Health Nurs. 2024. PMID: 38230967
References
Publication types
LinkOut - more resources
Full Text Sources