

Phylogenetic and functional diversity among Drosophila-associated metagenome-assembled genomes
- PMID: 40600712
- PMCID: PMC12282196
- DOI: 10.1128/msystems.00027-25
Phylogenetic and functional diversity among Drosophila-associated metagenome-assembled genomes
Abstract
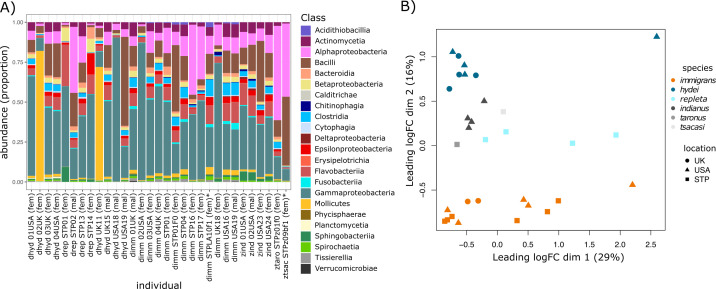
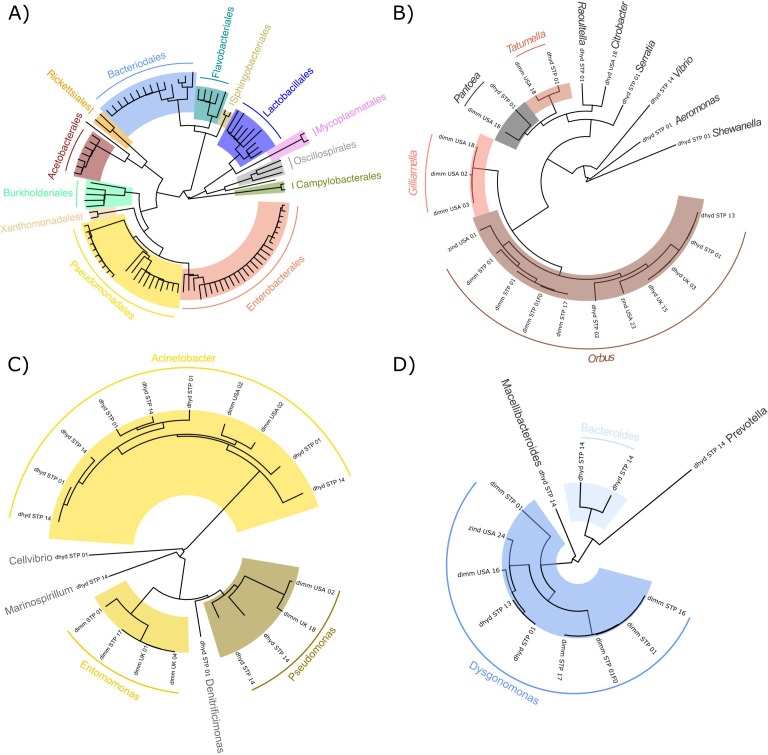
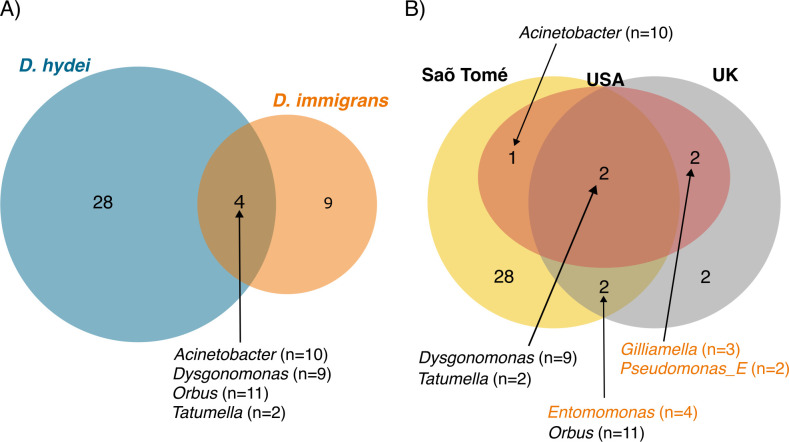
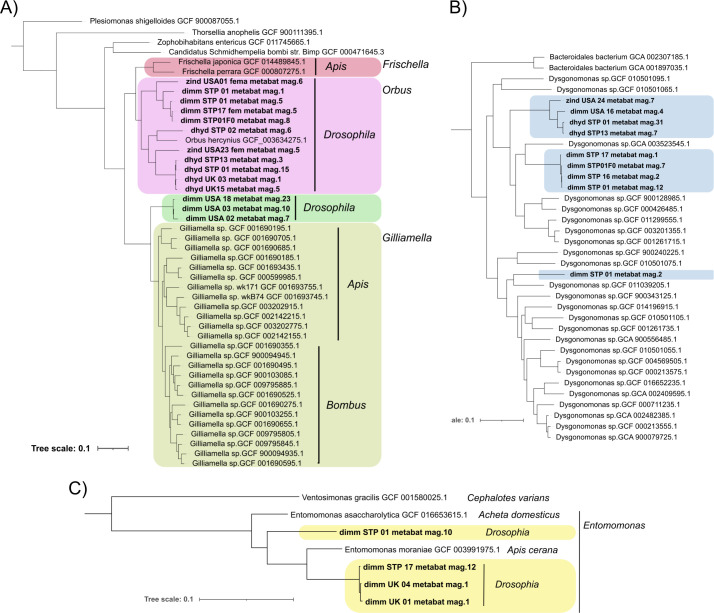
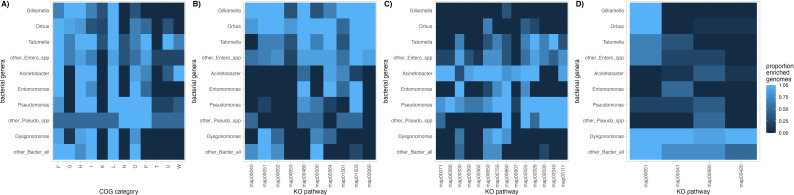
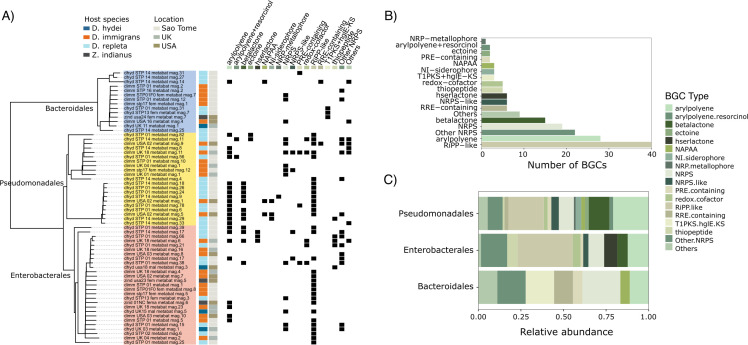
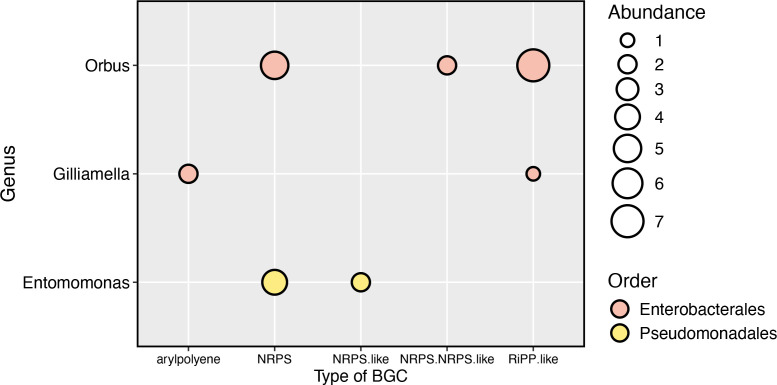
Host-associated microbial communities can mediate interactions between their hosts and biotic and abiotic environments. While much work has been done to document how microbiomes vary across species and environments, much less is known about the functional consequences of this variation. Here, we test for functional variation among drosophilid-associated bacteria by conducting Oxford Nanopore long-read sequencing and generating metagenome-assembled genomes (MAGs) from communities associated with six species of drosophilid flies collected from "anthropogenic" environments in North America, Europe, and Africa. Using phylogenetic analyses, we find that drosophilid flies harbor a diverse microbiome that includes core members closely related to the genera Gilliamella, Orbus, Entomomonas, Dysgonomonas, and others. Comparisons with publicly available bacterial genomes show that many of these genera are associated with phylogenetically diverse insect gut microbiomes. Using functional annotations and predicted secondary metabolite biosynthetic gene clusters, we show that MAGs belonging to different bacterial orders and genera vary in gene content and predicted functions, including metabolic capacity and how they respond to environmental stressors. Our results provide evidence that wild drosophilid flies harbor phylogenetically and functionally diverse microbial communities. These findings highlight a need to quantify the abundance and function of insect-associated bacteria from the genera Gilliamella, Orbus, Entomomonas, and others on the performance of their insect hosts across diverse environments.IMPORTANCEWhile much attention has been given to catalogue the taxonomic diversity intrinsic to host-associated microbiomes, much less is known about the functional consequences of this variation, especially in wild, non-model host species. In this study, we use long-read sequencing to generate and analyze 103 high-quality metagenome-assembled genomes from host-associated bacterial communities from six species of wild fruit fly (Drosophila). We find that the genomes of drosophilid-associated bacteria possess diverse metabolic pathways and biosynthetic gene clusters that are predicted to generate metabolites involved in nutrition and disease resistance, among other functions. Using functional gene predictions, we show that different bacterial lineages that comprise the insect microbiome differ in predicted functional capacities. Our findings highlight the functional variation intrinsic to microbial communities of wild insects and provide a step towards disentangling the ecological and evolutionary processes driving host-microbe symbioses.
Keywords: Drosophila; MAGs; Orbaceae; insects; metagenomics; microbiome; symbiosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ferguson LV, Dhakal P, Lebenzon JE, Heinrichs DE, Bucking C, Sinclair BJ. 2018. Seasonal shifts in the insect gut microbiome are concurrent with changes in cold tolerance and immunity. Funct Ecol 32:2357–2368. doi: 10.1111/1365-2435.13153 - DOI
-
- Maran AM, Weintraub MN, Pelini SL. 2020. Does stimulating ground arthropods enhance nutrient cycling in conventionally managed corn fields? Agric Ecosyst Environ 297:106934. doi: 10.1016/j.agee.2020.106934 - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
