

DNA-protein cross-links emerge as major contributors to chemotherapeutic cytotoxicity at physiological equitoxic doses
- PMID: 40604068
- PMCID: PMC12223252
- DOI: 10.1038/s41598-025-06705-3
DNA-protein cross-links emerge as major contributors to chemotherapeutic cytotoxicity at physiological equitoxic doses
Abstract
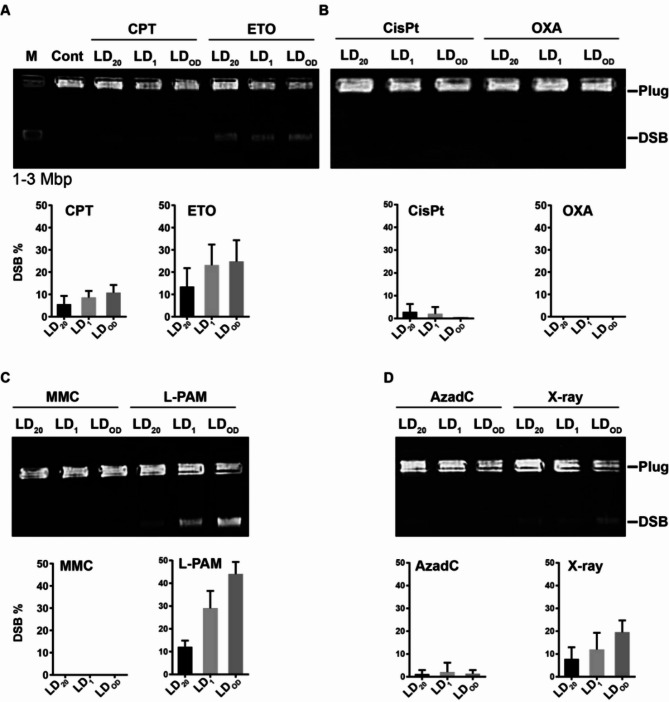
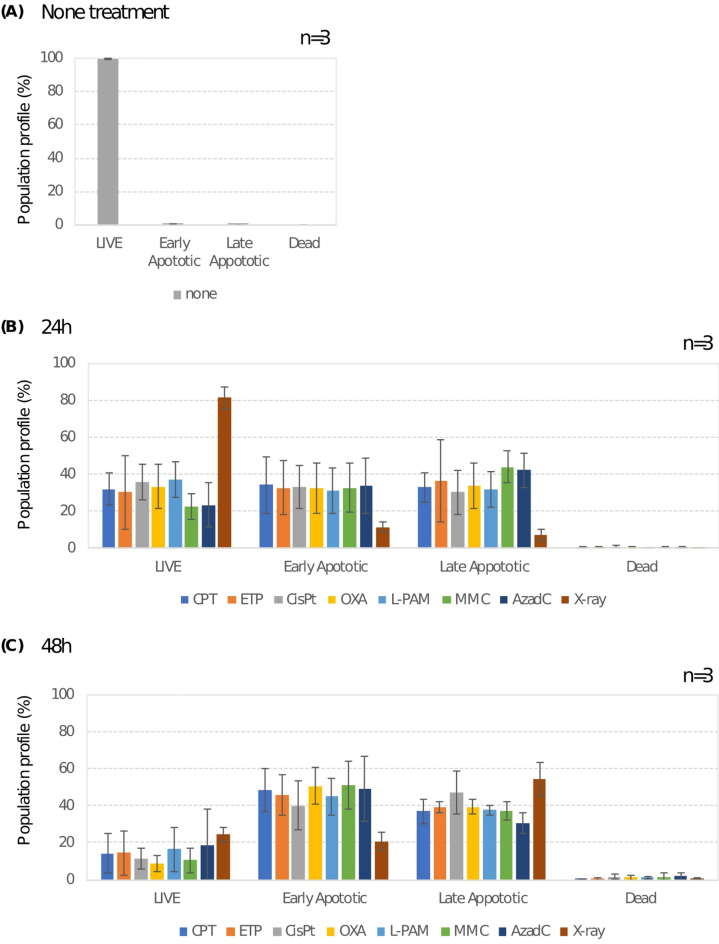
Chemotherapeutic drugs induce DNA damage, including double-strand breaks (DSBs), interstrand cross-links (ICLs), and DNA-protein cross-links (DPCs), to inhibit cancer cell proliferation. Understanding the relative contributions of these damages is essential for optimizing therapeutic strategies. To achieve physiologically relevant conditions, we determined the LD20 for four classes of chemotherapeutic agents and treated HeLa cells accordingly. Topoisomerase inhibitors (CPT, ETO) primarily induced DSBs and DPCs, whereas platinum-based agents (CisPt, OXA) predominantly caused DPCs and ICLs. The DNMT inhibitor AzadC was strongly associated with DPC formation. Although both L-PAM and MMC are bifunctional alkylating agents, their cytotoxic mechanisms differed; L-PAM induced DSBs, DPCs, and ICLs, while MMC primarily caused ICLs. DPCs were consistently detected across all drug treatments except MMC, with a half-life of 4.7 to 8.4 h, suggesting their prolonged impact on cytotoxicity. To assess apoptosis induction, we performed Annexin-V assays, which revealed significant apoptotic responses in all treated cells. CPT exhibited the highest proportion of early apoptotic cells (~ 80%) at 24 h, with all drug treatments shifting from early to late apoptosis over time. By 48 h, late apoptotic fractions exceeded 60% in CPT-, ETO-, and AzadC-treated cells. These findings highlight the critical role of DPCs in chemotherapeutic cytotoxicity and suggest that targeting apoptotic pathways could enhance cancer treatment efficacy.
Keywords: Cancer therapies; Chemotherapeutic drugs; Cytotoxicity; DNA damage; Equitoxic dose.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures




References
-
- Helleday, T., Petermann, E., Lundin, C., Hodgson, B. & Sharma, R. A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer. 8, 193–204 (2008). - PubMed
-
- McHugh, P. J., Spanswick, V. J. & Hartley, J. A. Repair of DNA interstrand cross-links: molecular mechanisms and clinical relevance. Lancet Oncol.2, 483–490 (2001). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
