

Mechanisms of delayed ischemia/reperfusion evoked ROS generation in the hippocampal CA1 zone of adult mouse brain slices
- PMID: 40604107
- PMCID: PMC12222531
- DOI: 10.1038/s41598-025-07070-x
Mechanisms of delayed ischemia/reperfusion evoked ROS generation in the hippocampal CA1 zone of adult mouse brain slices
Abstract
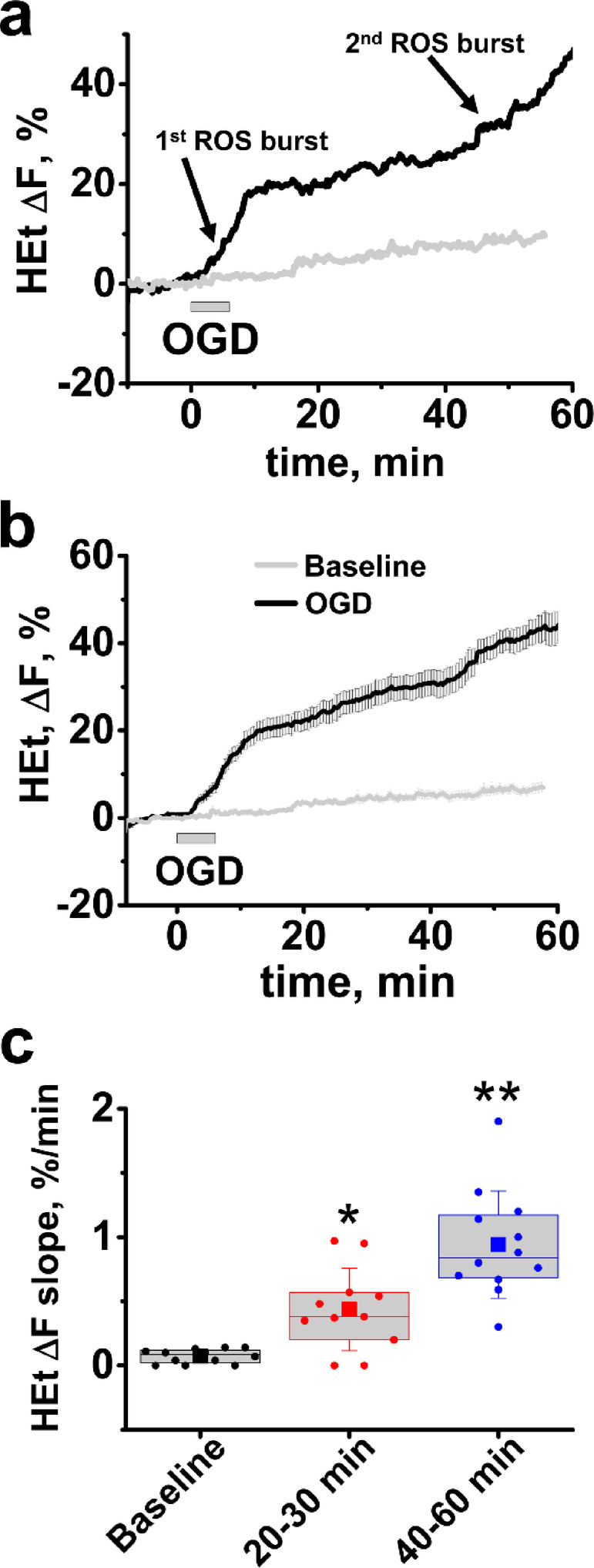
ROS overproduction is an important contributor to delayed ischemia/reperfusion induced neuronal injury, but relevant mechanisms remain poorly understood. We used oxygen-glucose deprivation (OGD)/reperfusion in mouse hippocampal slices to investigate ROS production in the CA1 pyramidal cell layer during and after transient ischemia. OGD evoked a 2-stage increase in ROS production: 1st-an abrupt increase in ROS generation starting during OGD followed by a marked slowing; and 2nd-a sharp ROS burst starting ~ 40 min after reperfusion. We further found that a slight mitochondrial hyperpolarization occurs shortly after OGD termination. Consequently, we showed that administration of low dose FCCP or of FTY720 (both of which cause mild, ~ 10%, mitochondrial depolarization), markedly diminished the delayed ROS burst, suggesting that mitochondrial hyperpolarization contributes to ROS production after reperfusion. Zn2+ chelation after OGD withdrawal also substantially decreased the late surge of ROS generation-in line with our prior studies indicating a critical contribution of Zn2+ entry into mitochondria via the mitochondrial Ca2+ uniporter (MCU) to mitochondrial damage after OGD. Thus, reperfusion-induced mitochondria hyperpolarization and mitochondrial Zn2+ accumulation both contribute to mitochondrial ROS overproduction after ischemia. As these events occur after reperfusion, they may be amenable to therapeutic interventions.
Keywords: Hippocampal slice; MCU; Mitochondria; Mitochondrial hyperpolarization; Oxygen glucose deprivation; Zn2+.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures




Update of
-
Mechanisms of delayed ischemia/reperfusion evoked ROS generation in the hippocampal CA1 zone of adult mouse brain slices.Res Sq [Preprint]. 2025 Apr 22:rs.3.rs-5640324. doi: 10.21203/rs.3.rs-5640324/v1. Res Sq. 2025. Update in: Sci Rep. 2025 Jul 2;15(1):23439. doi: 10.1038/s41598-025-07070-x. PMID: 40313775 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
