

Restoring p53 wild-type conformation in TP53-Y220C-mutant acute myeloid leukemia
- PMID: 40608889
- PMCID: PMC12320512
- DOI: 10.1182/blood.2025028935
Restoring p53 wild-type conformation in TP53-Y220C-mutant acute myeloid leukemia
Abstract
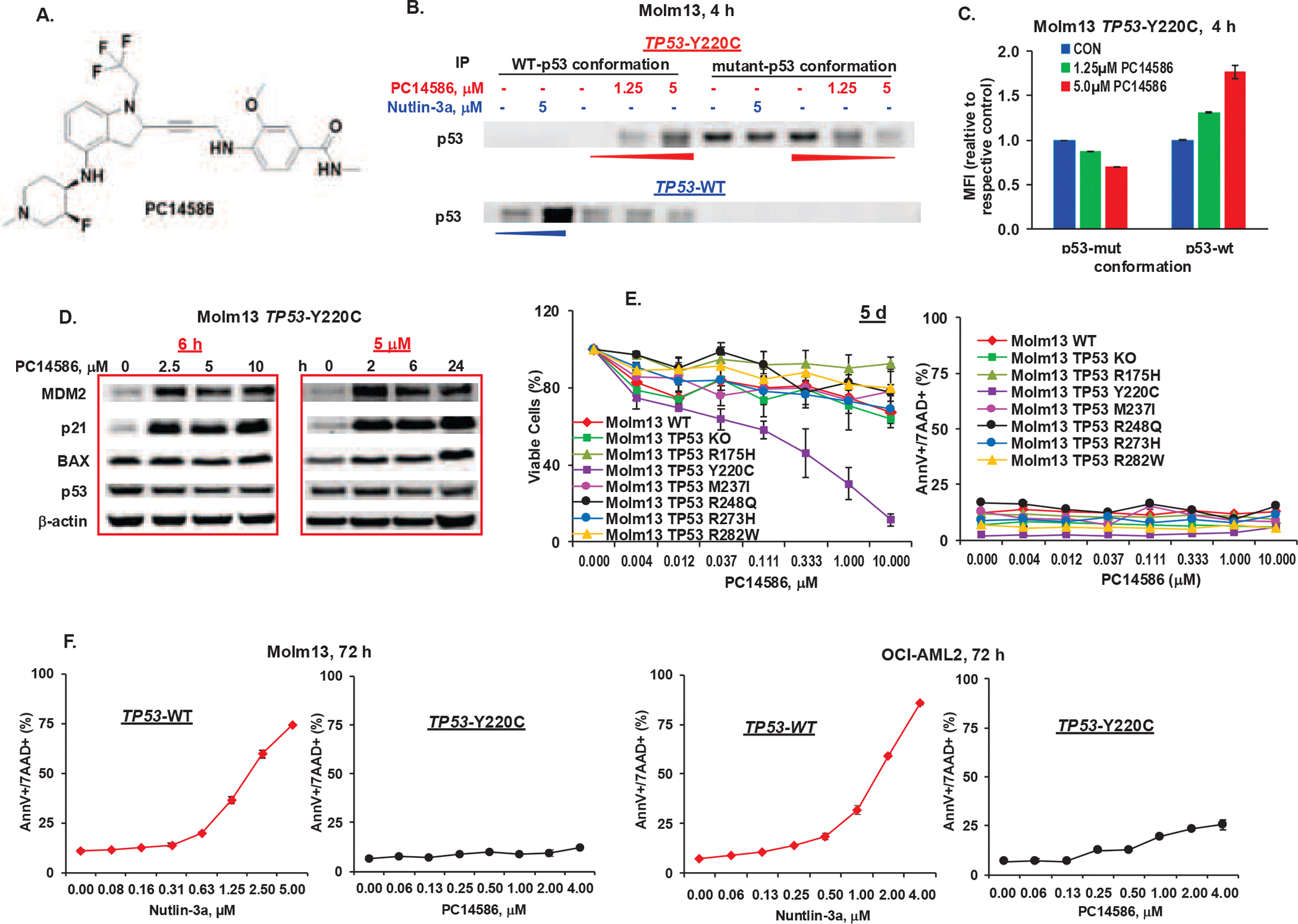
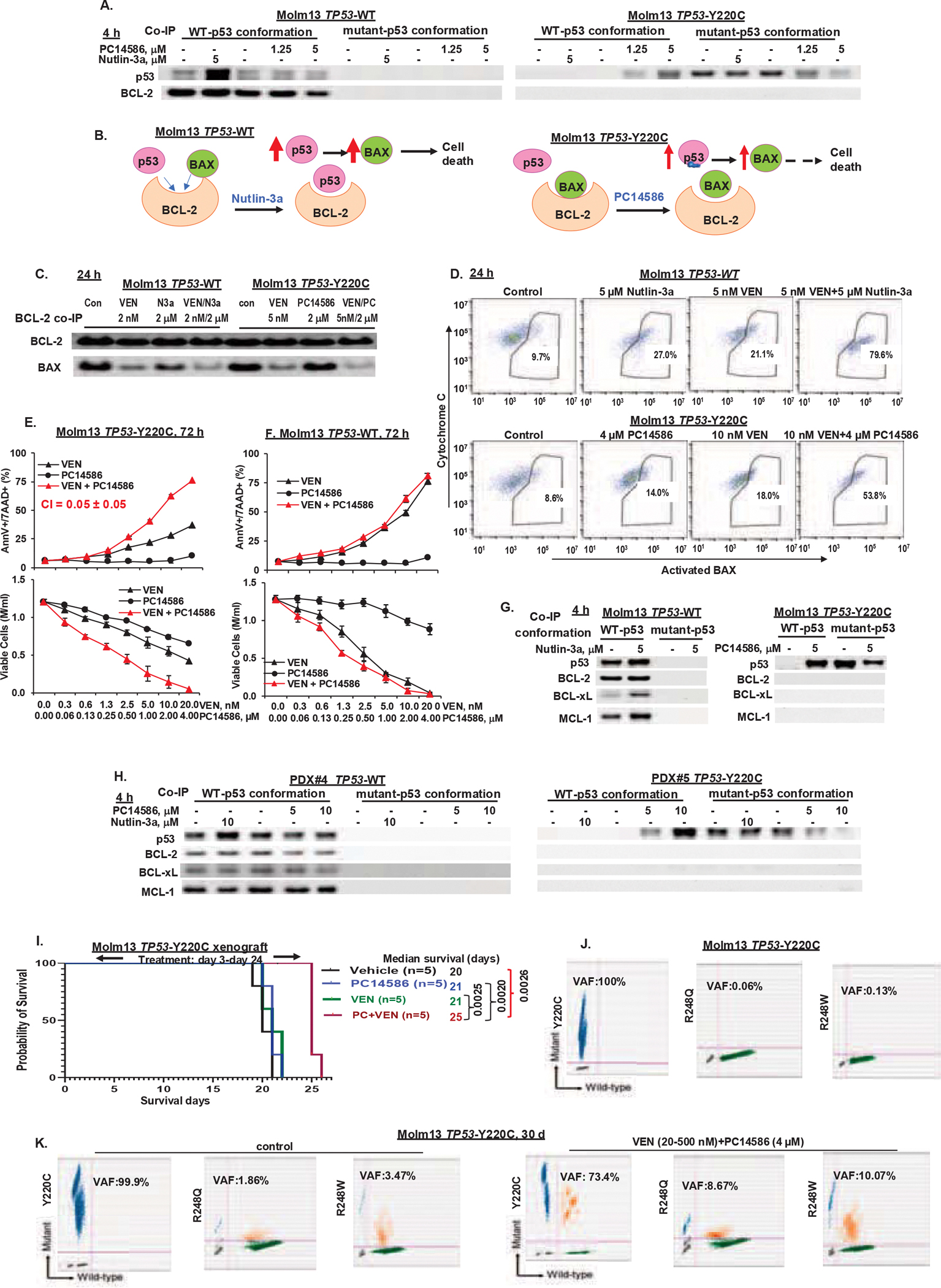
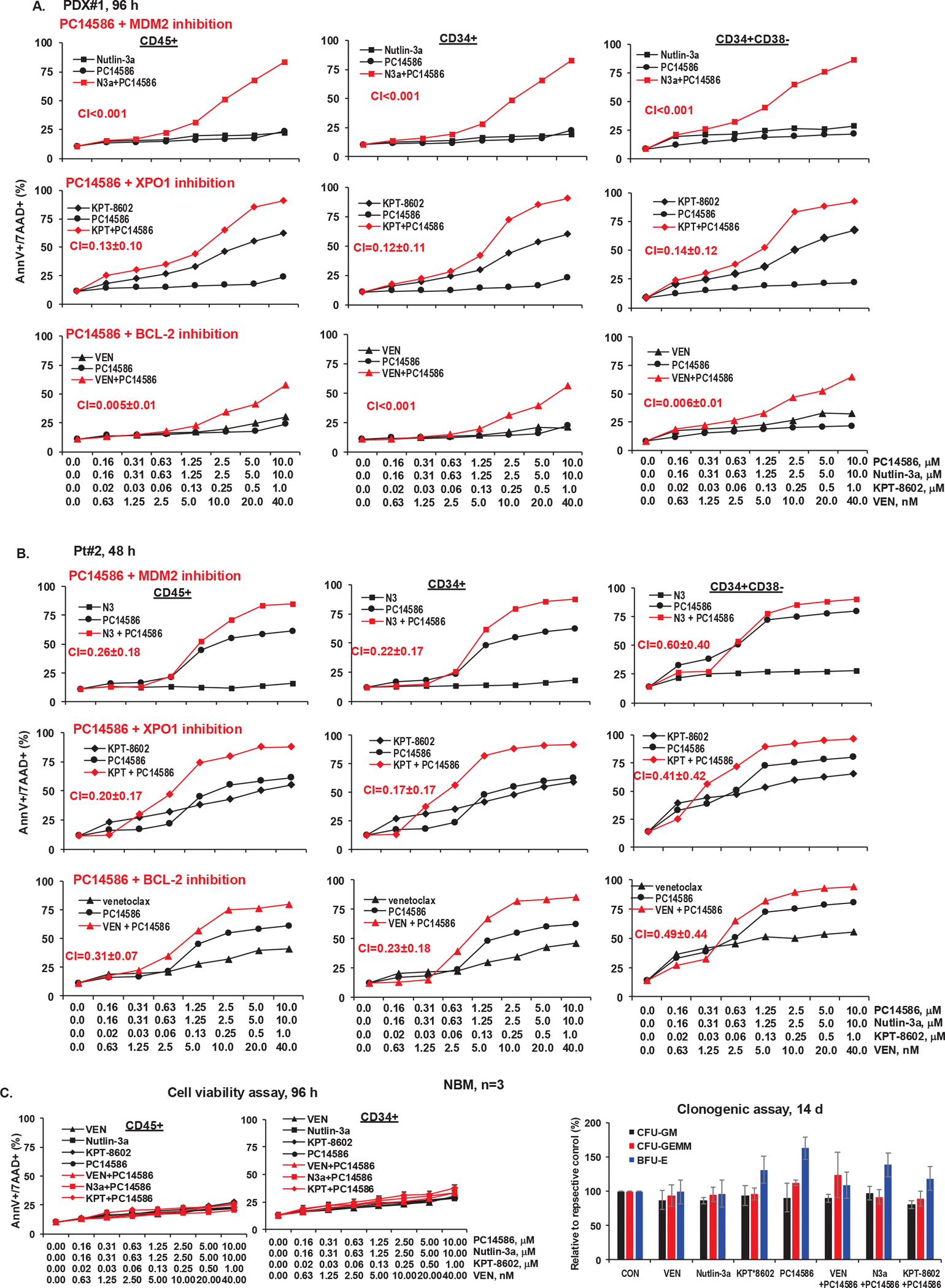
TP53-Y220C is a recurrent hotspot mutation in cancers and leukemias. It is observed predominantly in acute myeloid leukemia (AML)/myelodysplastic syndromes among hematological malignancies and is associated with poor outcome. The mutation creates a structural pocket in the p53 protein. PC14586 (rezatapopt) is a small molecule designed to bind to this pocket and thus restore a p53-wild type (p53-WT) conformation. We demonstrate that PC14586 converts p53-Y220C into a p53-WT conformation and activates p53 transcriptional targets, but surprisingly induces limited/no apoptosis in TP53-Y220C AML. Mechanistically, MDM2 induced by PC14586-activated conformational p53-WT and the nuclear exporter XPO1 reduce the transcriptional activities of p53, which are fully restored by inhibition of MDM2 and/or XPO1. Importantly, p53-WT protein can bind to BCL-2, competing with BAX in the BH3 binding pocket of BCL-2 and also binds to BCL-xL and MCL-1. However, such binding by PC14586-activated conformational p53-WT is not detected. Pharmacological inhibition of the BCL-2/BAX interaction with venetoclax fully compensates for this deficiency, induces massive cell death in AML cells and stem/progenitor cells in vitro and prolongs survival of TP53-Y220C AML xenografts in vivo. Collectively, we identified transcription-dependent and -independent mechanisms that limit the apoptogenic activities of reactivated conformational p53-WT and suggest approaches to optimize apoptosis induction in TP53-mutant leukemia. A clinical trial of PC14586 in TP53-Y220C AML/myelodysplastic syndromes has recently been initiated (NCT06616636).
Copyright © 2025 American Society of Hematology.
Conflict of interest statement
Figures



Similar articles
-
Rezatapopt: A promising small-molecule "refolder" specific for TP53Y220C mutant tumors.Neoplasia. 2025 Sep;67:101201. doi: 10.1016/j.neo.2025.101201. Epub 2025 Jun 20. Neoplasia. 2025. PMID: 40543428 Free PMC article.
-
A systematic overview of chemotherapy effects in acute myeloid leukaemia.Acta Oncol. 2001;40(2-3):231-52. doi: 10.1080/02841860151116321. Acta Oncol. 2001. PMID: 11441935
-
Synergistic Activity of Combined FLT3-ITD and MDM2 Inhibition with Quizartinib and Milademetan in FLT3-ITD Mutant/TP53 Wild-type Acute Myeloid Leukemias.Clin Cancer Res. 2025 Jul 15;31(14):3033-3047. doi: 10.1158/1078-0432.CCR-24-2764. Clin Cancer Res. 2025. PMID: 40327322 Free PMC article. Clinical Trial.
-
Thrombopoietin mimetics for patients with myelodysplastic syndromes.Cochrane Database Syst Rev. 2017 Sep 30;9(9):CD009883. doi: 10.1002/14651858.CD009883.pub2. Cochrane Database Syst Rev. 2017. PMID: 28962071 Free PMC article.
-
Early identification of TP53 mutations and TP53 allelic state in myelodysplastic neoplasms and acute myeloid leukemia via point-of-care p53 immunohistochemistry.Cancer. 2025 Jul 1;131(13):e35950. doi: 10.1002/cncr.35950. Cancer. 2025. PMID: 40542737
References
-
- Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711. - PubMed
-
- DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617–629. - PubMed
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
