

Acevaltrate as a novel ferroptosis inducer with dual targets of PCBP1/2 and GPX4 in colorectal cancer
- PMID: 40619432
- PMCID: PMC12230175
- DOI: 10.1038/s41392-025-02296-7
Acevaltrate as a novel ferroptosis inducer with dual targets of PCBP1/2 and GPX4 in colorectal cancer
Abstract
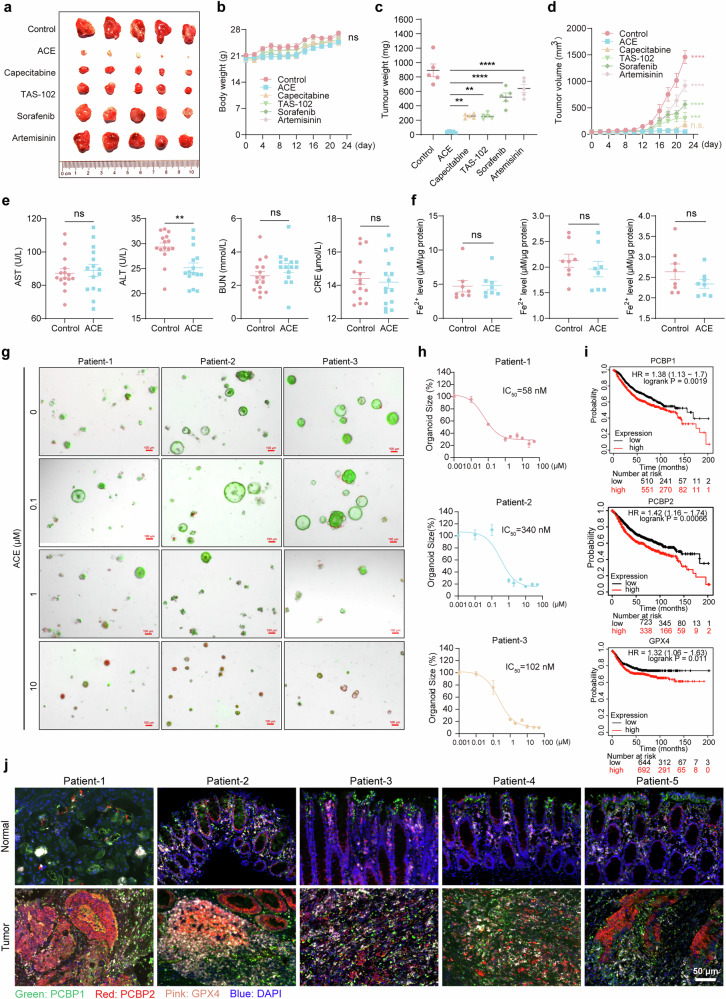
Ferroptosis induced by ferrous ions (Fe2+) and lipid peroxidation accumulation is a novel form of regulated cell death that has become a hot topic in tumor therapy research. Identifying small-molecule drugs that can induce ferroptosis in tumor cells is a very attractive therapeutic strategy. Here, we screened a natural product, acevaltrate (ACE), which rapidly and strongly induces ferroptosis in colorectal cancer cells. ACE not only increases Fe2+ levels in colorectal cancer cells by targeting iron chaperones PCBP1/2 and reducing their expression but also disrupts the antioxidant system of colorectal cancer cells by targeting GPX4 and inhibiting its enzymatic activity, leading to its ubiquitin-mediated degradation. This dual effect of ACE makes it significantly more effective than classical ferroptosis inducers in inducing ferroptosis. Our animal experiments revealed that the therapeutic effect of ACE surpasses that of established ferroptosis-inducing drugs and is superior to that of first-line clinical drugs such as capecitabine and TAS-102. Importantly, ACE also demonstrated superior inhibitory effects in colorectal tumor organoids versus at the cellular level, underscoring its potential for clinical application. This study pioneers the discovery of a small molecule inhibitor that targets both PCBP1/2 and GPX4, offering a novel therapeutic strategy for eliminating cancer cells through ferroptosis. Acevaltrate (ACE) was identified as a potent inducer of ferroptosis in colorectal cancer cells. ACE increases Fe2+ levels by targeting PCBP1/2 and disrupts the antioxidant system by inhibiting GPX4, leading to its degradation. This dual action makes ACE more effective at inducing ferroptosis than traditional inducers. Our study introduces ACE as the first small-molecule inhibitor of PCBP1/2 and GPX4, offering a new therapeutic approach for cancer cell elimination through ferroptosis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures








References
-
- Chen, X., Kang, R., Kroemer, G. & Tang, D. L. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol.18, 280–296 (2021). - PubMed
-
- Cheung, E. C. & Vousden, K. H. The role of ROS in tumour development and progression. Nat. Rev. Cancer22, 280–297 (2022). - PubMed
-
- Ai, Y. W., Meng, Y. T., Yan, B., Zhou, Q. Y. & Wang, X. D. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol. Cell84, 170–179 (2024). - PubMed
-
- Angeli, J. P. F., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer19, 405–414 (2019). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
