

Dynamicasome-a molecular dynamics-guided and AI-driven pathogenicity prediction catalogue for all genetic mutations
- PMID: 40624350
- PMCID: PMC12234709
- DOI: 10.1038/s42003-025-08334-y
Dynamicasome-a molecular dynamics-guided and AI-driven pathogenicity prediction catalogue for all genetic mutations
Abstract
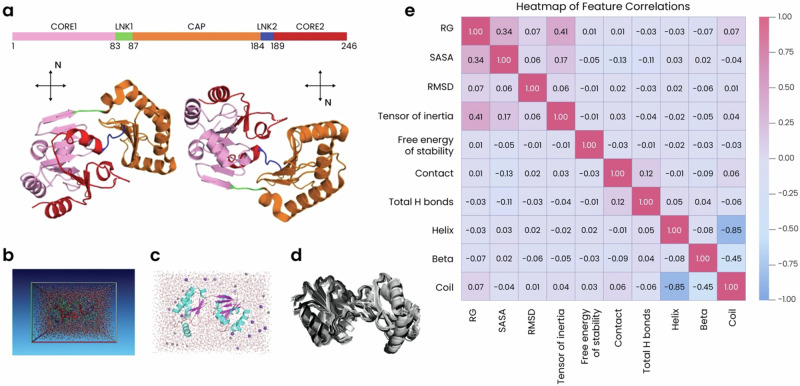
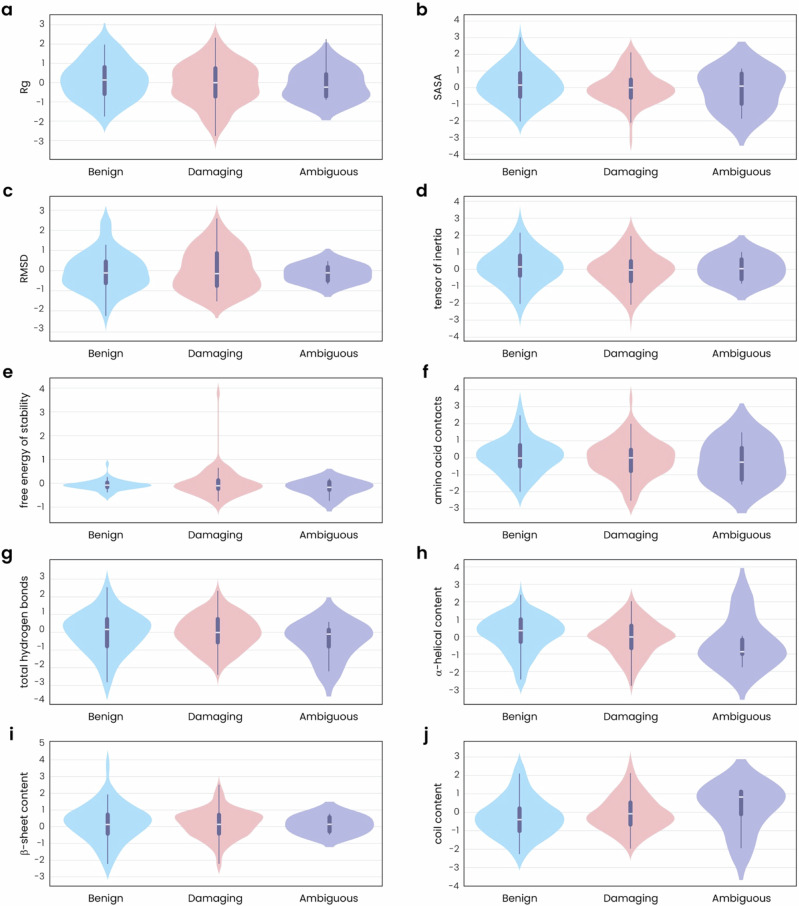
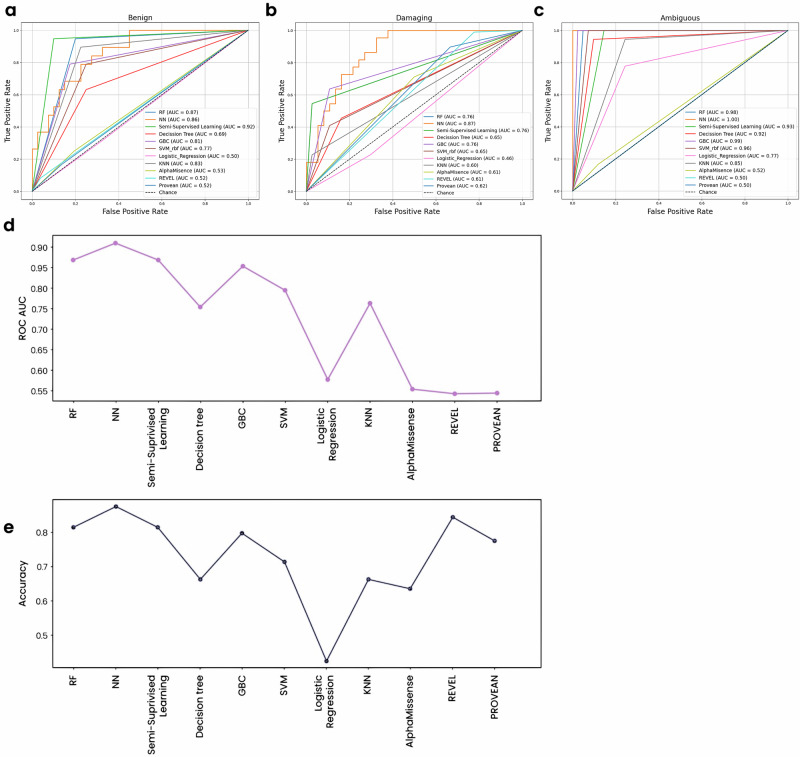
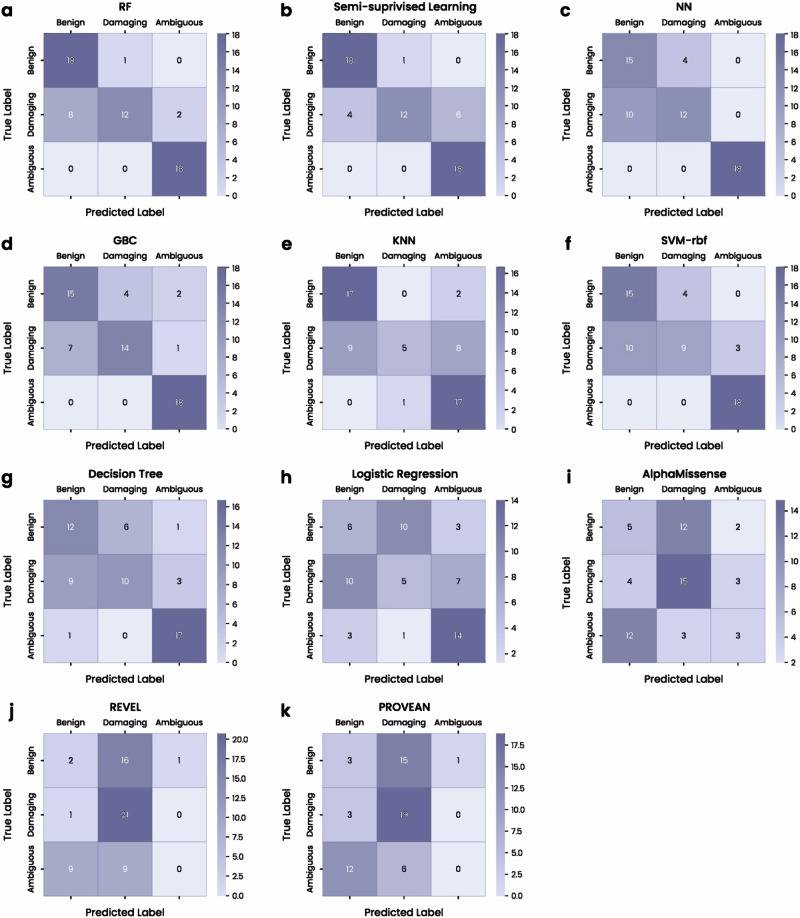
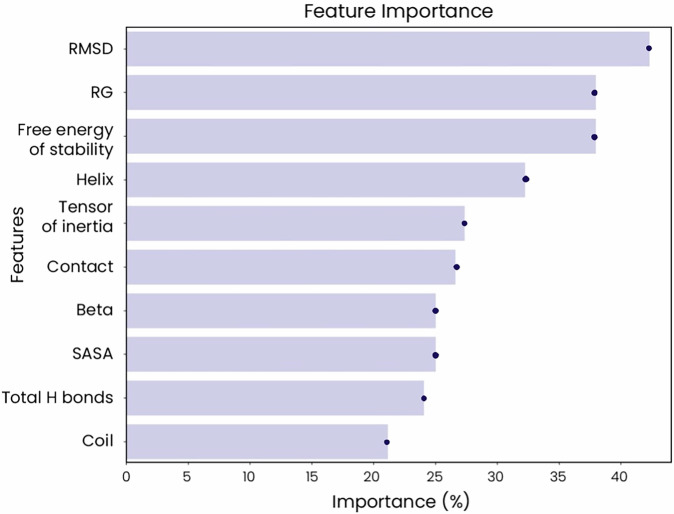
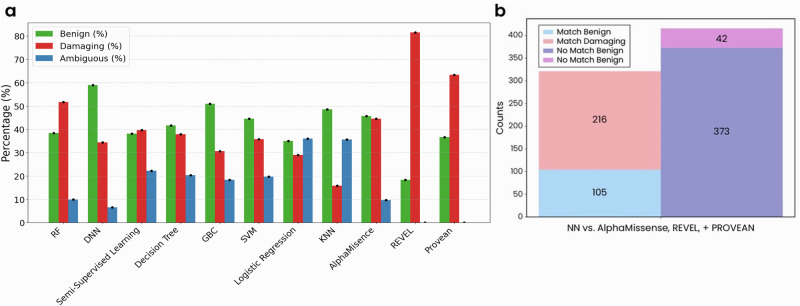
Advances in genomic medicine accelerate the identification of mutations in disease-associated genes, but the pathogenicity of many mutations remains unknown, hindering their use in diagnostics and clinical decision-making. Predictive AI models are generated to combat this issue, but current tools display low accuracy when tested against functionally validated datasets. We show that integrating detailed conformational data extracted from molecular dynamics simulations (MDS) into advanced AI-based models increases their predictive power. We carry out an exhaustive mutational analysis of the disease gene PMM2 and subject structural models of each variant to MDS. AI models trained on this dataset outperform existing tools when predicting the known pathogenicity of mutations. Our best performing model, a neuronal networks model, also predicts the pathogenicity of several PMM2 mutations currently considered of unknown significance. We believe this model helps alleviate the burden of unknown variants in genomic medicine.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Improving reliability of movement assessment in Parkinson's disease using computer vision-based automated severity estimation.J Parkinsons Dis. 2025 Mar;15(2):349-360. doi: 10.1177/1877718X241312605. Epub 2025 Feb 13. J Parkinsons Dis. 2025. PMID: 39973505
-
Artificial intelligence for detecting keratoconus.Cochrane Database Syst Rev. 2023 Nov 15;11(11):CD014911. doi: 10.1002/14651858.CD014911.pub2. Cochrane Database Syst Rev. 2023. PMID: 37965960 Free PMC article.
-
The comparative and added prognostic value of biomarkers to the Revised Cardiac Risk Index for preoperative prediction of major adverse cardiac events and all-cause mortality in patients who undergo noncardiac surgery.Cochrane Database Syst Rev. 2021 Dec 21;12(12):CD013139. doi: 10.1002/14651858.CD013139.pub2. Cochrane Database Syst Rev. 2021. PMID: 34931303 Free PMC article.
References
-
- OMIM. in Online Mendelian Inheritance in Man OMIM® (McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
