

Advancing CAR-based cell therapies for solid tumours: challenges, therapeutic strategies, and perspectives
- PMID: 40624498
- PMCID: PMC12232864
- DOI: 10.1186/s12943-025-02386-8
Advancing CAR-based cell therapies for solid tumours: challenges, therapeutic strategies, and perspectives
Abstract
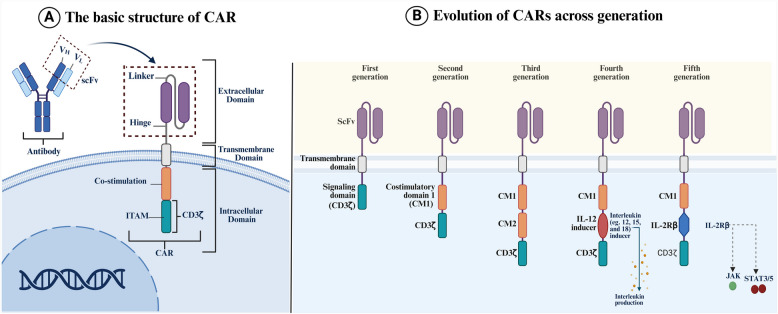
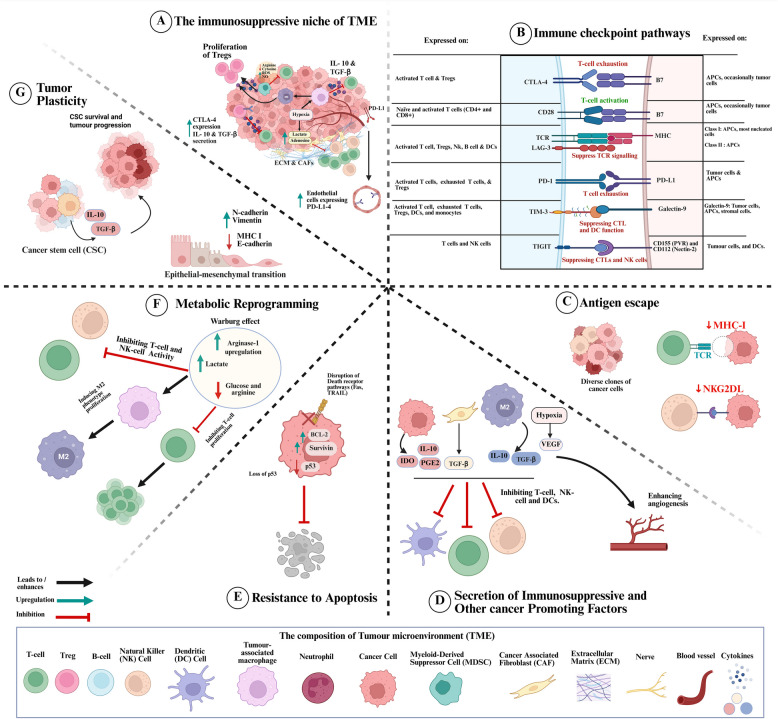
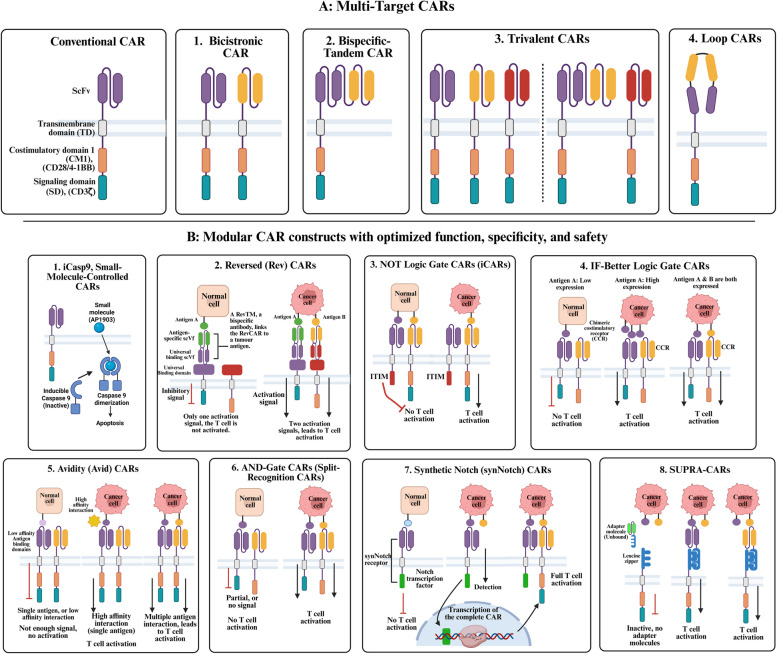
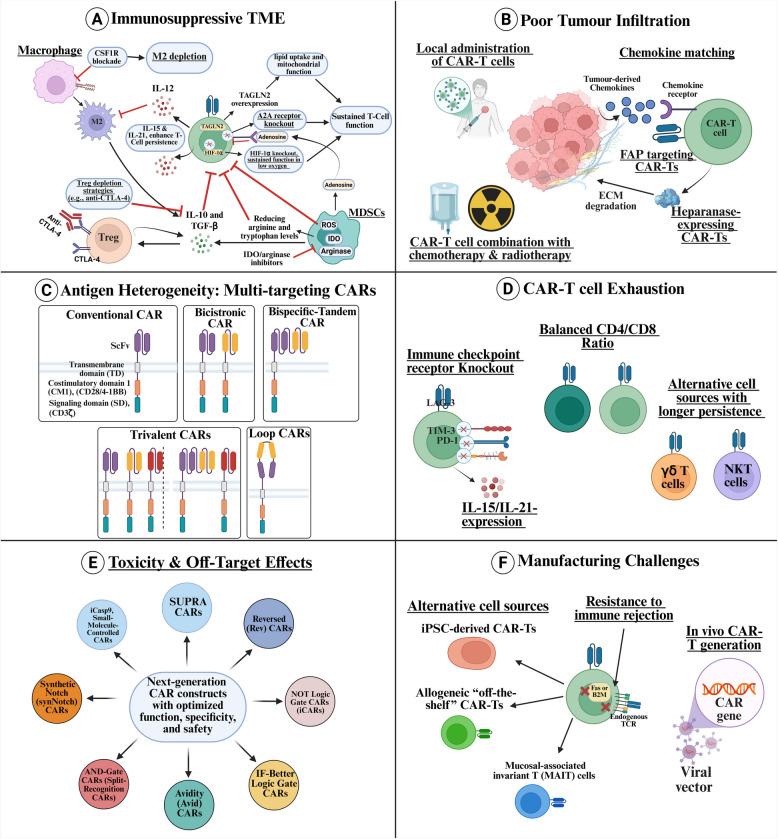
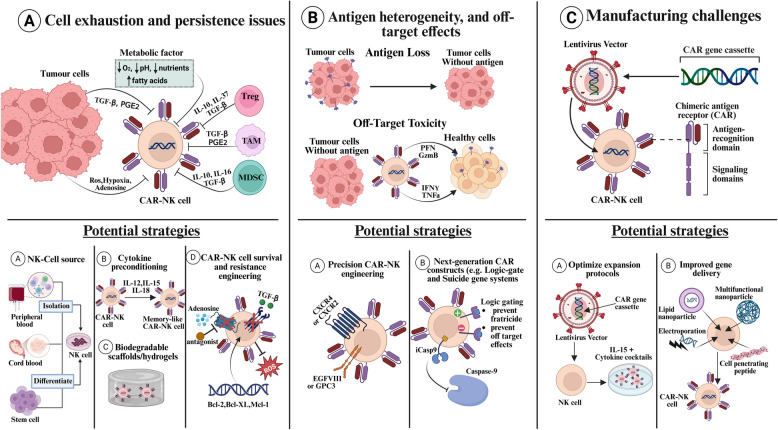
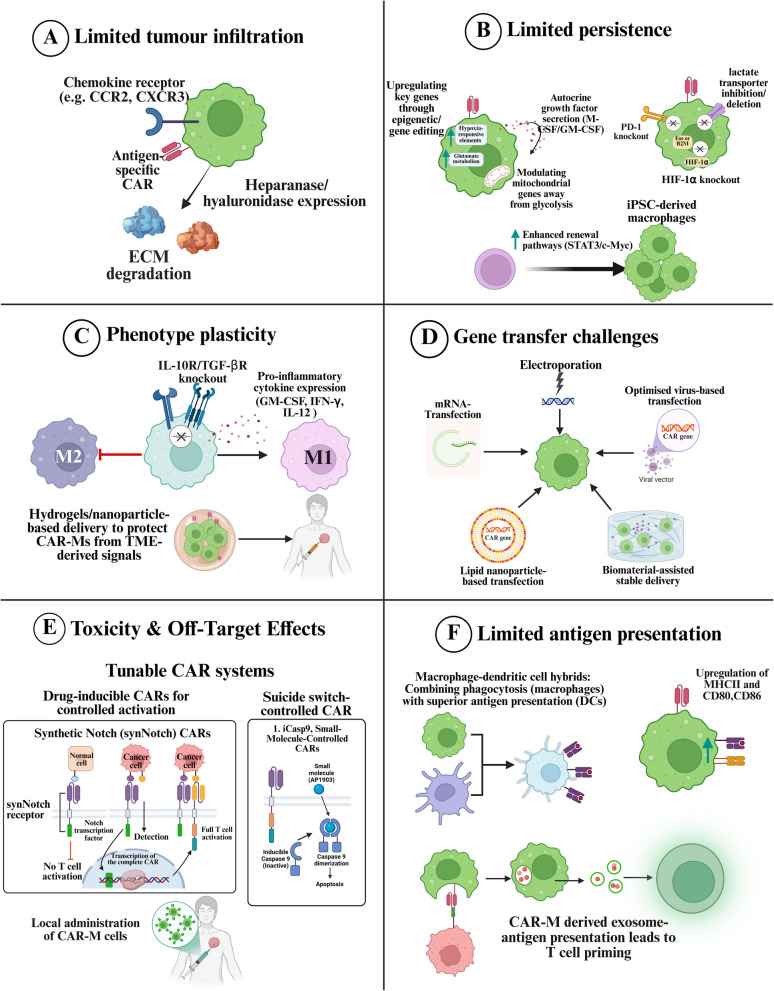
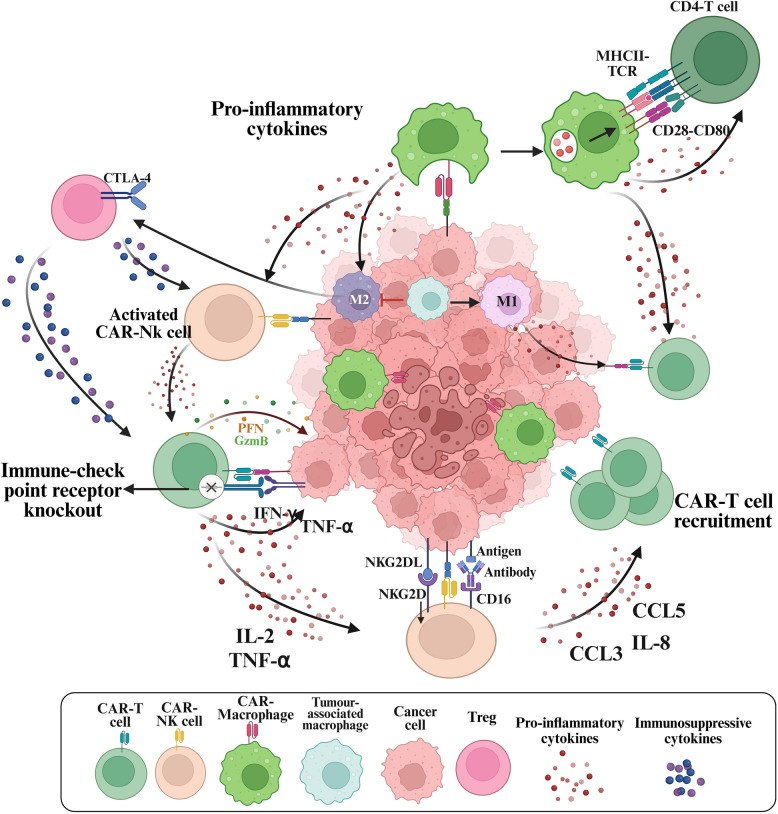
Chimeric antigen receptor-cell therapies have demonstrated remarkable success in haematological malignancies but face significant hurdles in solid tumours. The hostile tumour microenvironment, antigen heterogeneity, limited tumour infiltration, and CAR-cell exhaustion contribute to reduced efficacy. Additionally, toxicity, off-target effects, and manufacturing challenges limit widespread clinical adoption. Overcoming these barriers requires a multifaceted approach that enhances CAR-cell persistence, trafficking, and tumour-specific targeting. Recent advancements in alternative cellular therapies, such as CAR-natural killer cells, CAR-macrophages, gamma delta CAR-T cells, and CAR-natural killer T cells, provide promising avenues for improving efficacy. These strategies leverage distinct immune cell properties to enhance tumour recognition and persistence. Furthermore, combination therapies, including chemotherapy, radiotherapy, antibodies, small molecule inhibitors, cancer vaccines, oncolytic viruses, and multi-CAR cell combination therapy, offer synergistic potential by modulating the TME and improving CAR-cell functionality. This review explores the challenges of CAR-based cellular therapies in solid tumours and highlights emerging strategies to overcome therapeutic limitations. By integrating novel cellular platforms and combination approaches, we seek to provide insights into optimising CAR-cell therapies for durable responses in solid malignancies.
Keywords: CAR-Macrophages; CAR-NK cells; CAR-T cells; Challenges; Combination therapy; Optimisation Strategies; Solid tumours.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: All authors consent to the publication. Competing interests: The authors declare no competing interests.
Figures
References
-
- Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63. - PubMed
-
- Montagna E, de Campos NSP, Porto VA, da Silva GCP, Suarez ER. CD19 CAR T cells for B cell malignancies: a systematic review and meta-analysis focused on clinical impacts of CAR structural domains, manufacturing conditions, cellular product, doses, patient’s age, and tumor types. BMC Cancer. 2024;24:1037. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
