

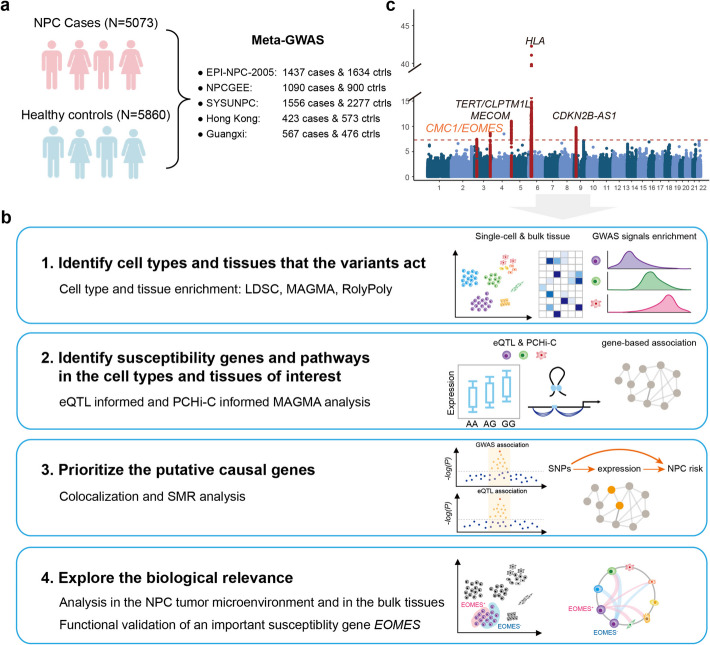
A genome-wide association study integrated with single-cell and bulk profiles uncovers susceptibility genes for nasopharyngeal carcinoma involved in tumorigenesis via regulation of T cells
- PMID: 40624598
- PMCID: PMC12236000
- DOI: 10.1186/s13059-025-03657-9
A genome-wide association study integrated with single-cell and bulk profiles uncovers susceptibility genes for nasopharyngeal carcinoma involved in tumorigenesis via regulation of T cells
Abstract
Background: Nasopharyngeal carcinoma is an aggressive malignancy originating from the nasopharyngeal mucosa and associated with genetic factors. Many nasopharyngeal carcinoma susceptibility loci have been identified by genome-wide association studies (GWASs), but their underlying functional insights are largely unexplained.
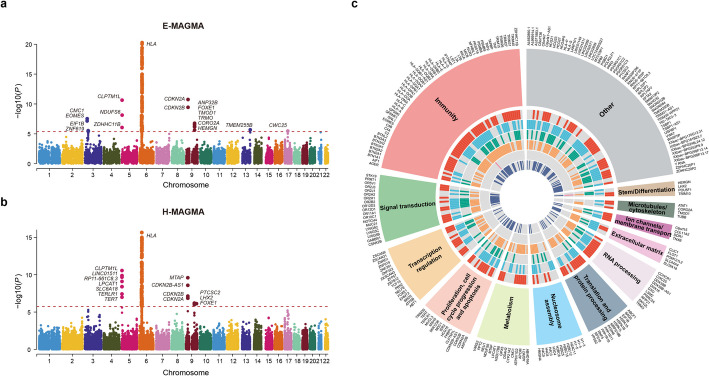
Results: A meta-GWAS including 5073 nasopharyngeal carcinoma patients and 5860 controls from nasopharyngeal carcinoma endemic areas identifies a total of 863 significant SNPs, including SNPs at a novel locus 3p24.1 (rs56365817; nearby genes: CMC1/EOMES). By integrating the GWAS signals with single-cell and bulk profiles, we find nasopharyngeal carcinoma susceptibility robustly associated with T cells in different methods and datasets. In nasopharyngeal carcinoma-associated cell type, we identify 234 putative susceptibility genes (81.62% of them novel), mainly enriched in immune-related biological processes. Five putative causal genes are prioritized. We perform in-depth bioinformatic analysis and functional experiments for EOMES, finding that the nasopharyngeal carcinoma-risk alleles of four functional SNPs upregulate EOMES expression by promoting the activity of regulatory elements in T cells, and EOMES participates in nasopharyngeal carcinoma tumorigenesis via regulation of CD8+ T cell exhaustion in the tumor microenvironment.
Conclusions: This study uncovers novel nasopharyngeal carcinoma susceptibility genes and their functional cell types, which improves the understanding of nasopharyngeal carcinoma genetic etiology.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Written informed consent was obtained from all patients. This study was approved by the Institutional Review Boards of Sun Yat-sen University Cancer Center. Competing interests: The authors declare no competing interests.
Figures





References
MeSH terms
Substances
Grants and funding
- 82373656/the National Natural Science Foundation of China
- 82473703/the National Natural Science Foundation of China
- 82404339/the National Natural Science Foundation of China
- 82273705/the National Natural Science Foundation of China
- 2024A04J4560/the Science and Technology Planning Project of Guangzhou, China
- 2024A04J00693/the Science and Technology Planning Project of Guangzhou, China
- QT2024-030/the Young Talent Support Project of Guangzhou Association for Science and Technology
- 24ykqb002/the Fundamental Research Funds for the Central Universities, Sun Yat-sen University
- 24qnpy292/the Fundamental Research Funds for the Central Universities, Sun Yat-sen University
- YTP-SYSUCC-0076/the Young Talents Program of Sun Yat-sen University Cancer Center
- YTP-SYSUCC-0081/the Young Talents Program of Sun Yat-sen University Cancer Center
- YSTTGDPMAA202502/Young Science and Technology Talent Support Program of Guangdong Precision Medicine Application Association
- 2023ZD0501000/Noncommunicable Chronic Diseases-National Science and Technology Major Project
- CIRP-SYSUCC-0017/Cancer Innovative Research Program of Sun Yat-sen University Cancer Center
LinkOut - more resources
Full Text Sources
Research Materials
