

BIK polymorphism and proteasome regulation unveil host risk factor for severe influenza
- PMID: 40627391
- PMCID: PMC12280918
- DOI: 10.1073/pnas.2424367122
BIK polymorphism and proteasome regulation unveil host risk factor for severe influenza
Erratum in
-
Correction for Soni et al., BIK polymorphism and proteasome regulation unveil host risk factor for severe influenza.Proc Natl Acad Sci U S A. 2025 Sep 2;122(35):e2521789122. doi: 10.1073/pnas.2521789122. Epub 2025 Aug 28. Proc Natl Acad Sci U S A. 2025. PMID: 40875814 Free PMC article. No abstract available.
Abstract
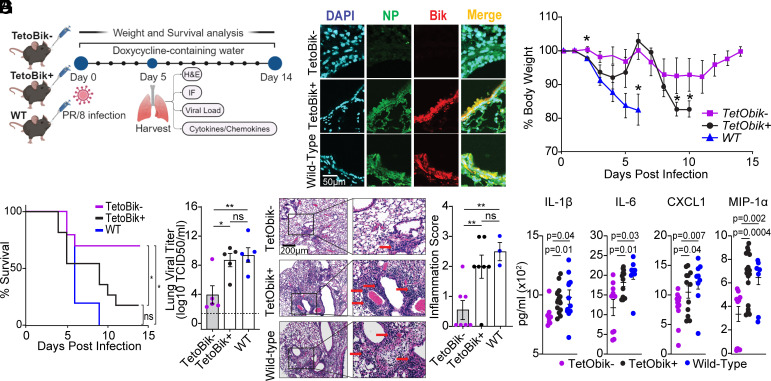
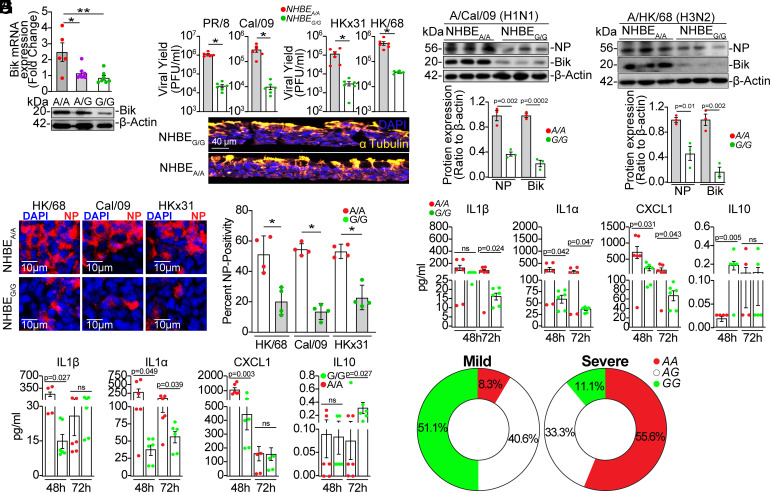
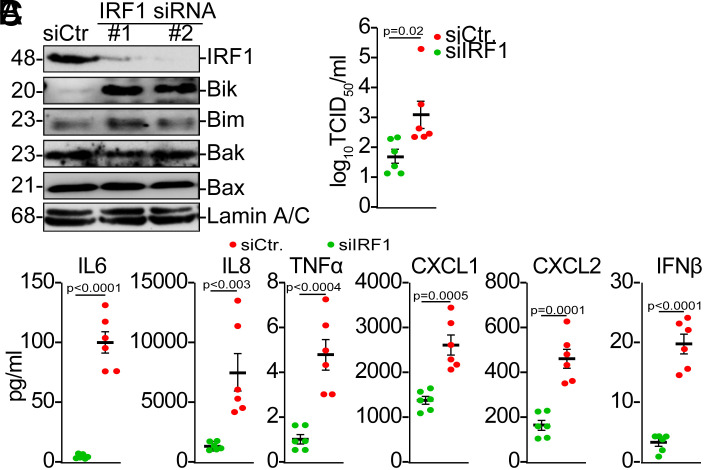
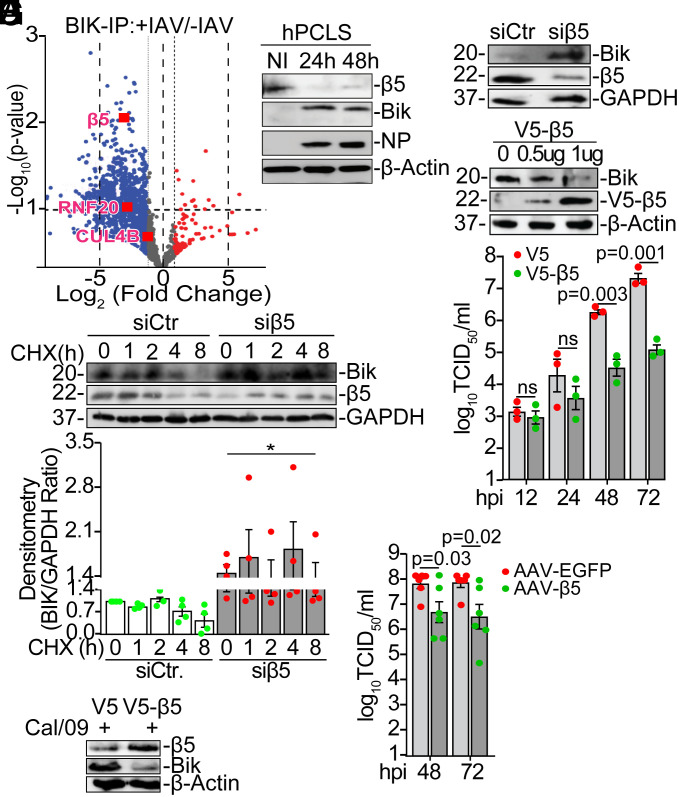
Influenza A viruses (IAVs) pose a significant public health threat, with host factors playing a crucial role in disease severity. We investigated the role of Bcl-2-interacting killer (BIK) in IAV infection using cellular and mouse models, and influenza-infected human cohort. In airway epithelial cells (AECs), BIK deficiency impaired viral replication, while BIK restoration enhanced it. Conversely, airway-specific BIK overexpression in mice increased viral load, inflammation, and mortality, whereas BIK suppression conferred protection. Critically, a genetic variation (rs738276) in the BIK gene, influencing BIK expression, correlates with altered viral replication in air-liquid interface differentiated primary normal human bronchial epithelial cells and influenza severity in humans. Mechanistically, we demonstrate that IAV nucleoprotein (NP) suppresses β5, a subunit of the proteasome, leading to increased BIK levels and enhanced viral replication. Conversely, β5 treatment dampened BIK levels and protected mice from IAV-induced morbidity and mortality. Furthermore, BIK interacts with NP, disrupting the Bcl-2/NP interaction and promoting viral replication. Our findings uncover an IAV-BIK-β5 axis that governs viral replication, suggesting that targeting BIK or β5 may offer therapeutic strategies against influenza.
Keywords: BIK; Beta 5; Influenza A virus; Proteasome; SNP.
Conflict of interest statement
Competing interests statement:The A.G.-S. laboratory has received research support from GSK, Pfizer, Senhwa Biosciences, Kenall Manufacturing, Blade Therapeutics, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N-fold LLC, Model Medicines, Atea Pharma, Applied Biological Laboratories and Merck, outside of the reported work. A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Castlevax, Amovir, Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Pagoda, Accurius, Esperovax, Applied Biological Laboratories, Pharmamar, CureLab Oncology, CureLab Veterinary, Synairgen, Paratus, Pfizer and Prosetta, outside of the reported work. A.G.-S. has been an invited speaker in meeting events organized by Seqirus, Janssen, Abbott, Astrazeneca and Novavax. A.G.-S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work. Y.T. has received research funds from Bayer Pharmaceuticals, AstraZeneca, and Verra Therapeutics. P.G.T. is on the SAB of Immunoscape and Shennon Bio, has received research support and personal fees from Elevate Bio, and consulted for or had travel expenses paid by 10x Genomics, Illumina, Pfizer, Cytoagents, Merck, Bluebird, and JNJ. S.S., S.Y., E.K.A., H.P., M.P., S.E.Z., L.R., V.A., L.A., R.S.N., A.L.M., J.C.H, M.R., Y.T., and Y.A.M. have declared that no conflicts of interest exist. R.A.M. served as a scientific advisor for Valneva SE.
Figures




References
-
- Fiore A. E., et al. , Prevention and control of influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2008. MMWR Recomm. Rep. 57, 1–60 (2008). - PubMed
-
- Molinari N. A., et al. , The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 25, 5086–5096 (2007). - PubMed
-
- Davies W. L., et al. , Antiviral activity of 1-adamantanamine (amantadine). Science 144, 862–863 (1964). - PubMed
MeSH terms
Substances
Grants and funding
- R01 HL171164/HL/NHLBI NIH HHS/United States
- R01 AI154470/AI/NIAID NIH HHS/United States
- R01AI148180/HHS | National Institutes of Health (NIH)
- R01 HL140839/HL/NHLBI NIH HHS/United States
- 75N93021C00016/AI/NIAID NIH HHS/United States
- U01 AI187057/AI/NIAID NIH HHS/United States
- RO1HL068111 R01HL140839/HHS | National Institutes of Health (NIH)
- R01 HL068111/HL/NHLBI NIH HHS/United States
- P30 CA016058/CA/NCI NIH HHS/United States
- 75N93021C00014/AI/NIAID NIH HHS/United States
- R01AI154470/HHS | National Institutes of Health (NIH)
- R01 AI148180/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
