

Haploblocks contribute to parallel climate adaptation following global invasion of a cosmopolitan plant
- PMID: 40629086
- PMCID: PMC12328208
- DOI: 10.1038/s41559-025-02751-2
Haploblocks contribute to parallel climate adaptation following global invasion of a cosmopolitan plant
Abstract
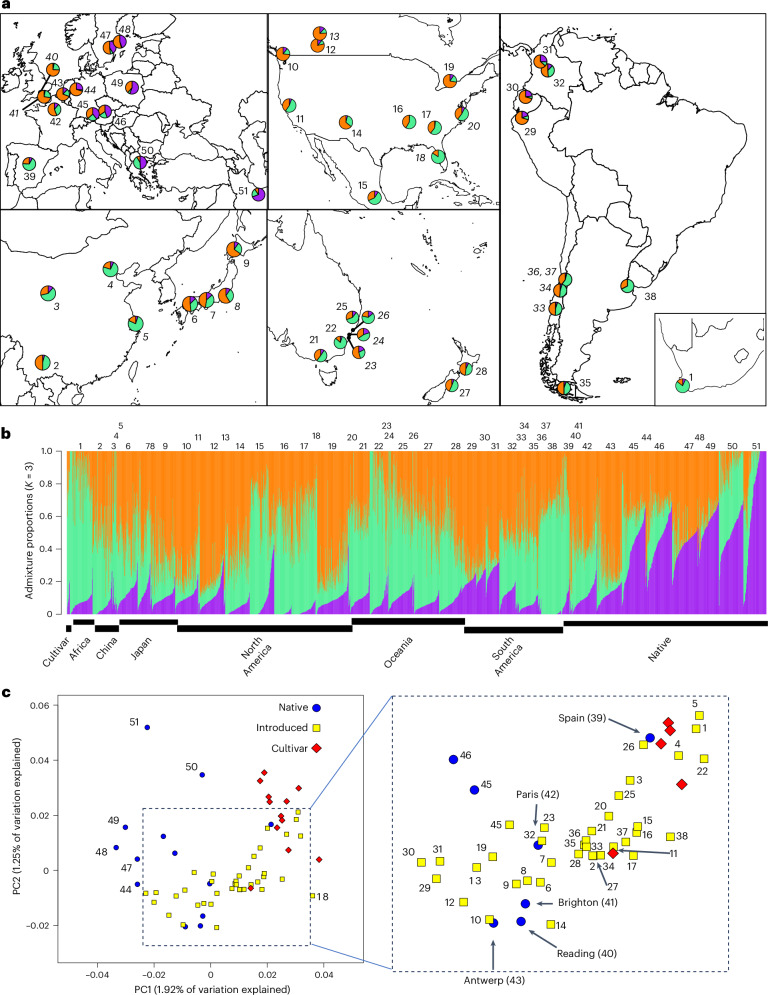
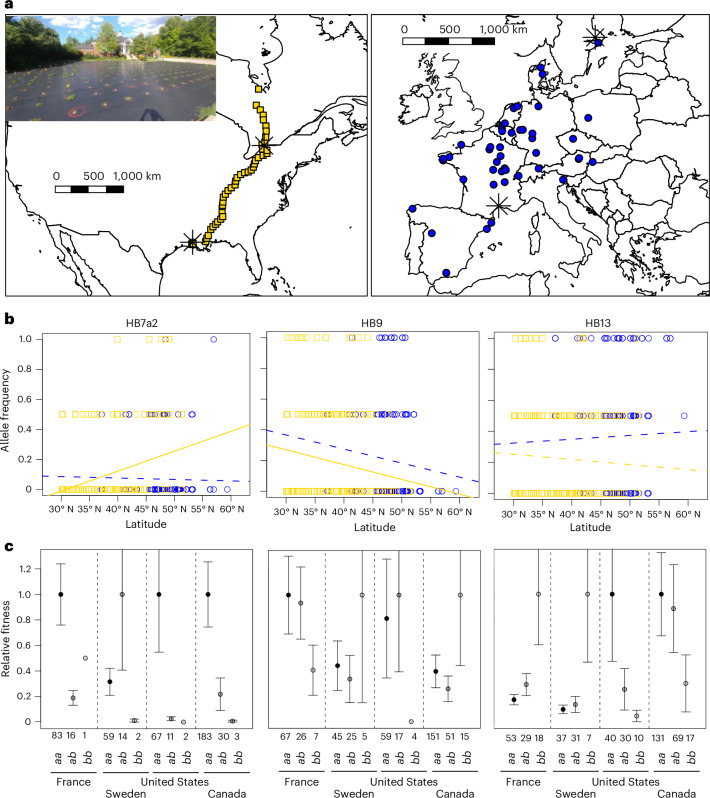
The role of rapid adaptation during species invasions has historically been minimized with the assumption that introductions consist of few colonists and limited genetic diversity. While overwhelming evidence suggests that rapid adaptation is more prevalent than originally assumed, the demographic and adaptive processes underlying successful invasions remain unresolved. Here we leverage a large whole-genome sequence dataset to investigate the relative roles of colonization history and adaptation during the worldwide invasion of the forage crop, Trifolium repens (Fabaceae). We show that introduced populations encompass high levels of genetic variation with little evidence of bottlenecks. Independent colonization histories on different continents are evident from genome-wide population structure. Five haploblocks-large haplotypes with limited recombination-on three chromosomes exist as standing genetic variation within the native and introduced ranges and exhibit strong signatures of parallel climate-associated adaptation across continents. Field experiments in the native and introduced ranges demonstrate that three of the haploblocks strongly affect fitness and exhibit patterns of selection consistent with local adaptation across each range. Our results provide strong evidence that large-effect structural variants contribute substantially to rapid and parallel adaptation of an introduced species throughout the world.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













References
-
- Pimentel, D., Lach, L., Zuniga, R. & Morrison, D. Environmental and economic costs of nonindigenous species in the United States. BioScience50, 53 (2000). - DOI
-
- Pimentel, D., Zuniga, R. & Morrison, D. Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol. Econ.52, 273–288 (2005). - DOI
-
- Hayes, K. R. & Barry, S. C. Are there any consistent predictors of invasion success? Biol. Invasions10, 483–506 (2008). - DOI
MeSH terms
Grants and funding
- OIA-1920858/National Science Foundation (NSF)
- DBI-2244712/National Science Foundation (NSF)
- EQPEQ 423691/Gouvernement du Canada | Natural Sciences and Engineering Research Council of Canada (Conseil de Recherches en Sciences Naturelles et en Génie du Canada)
- RGPIN 2016 06063/Gouvernement du Canada | Natural Sciences and Engineering Research Council of Canada (Conseil de Recherches en Sciences Naturelles et en Génie du Canada)
- RGP0001/Human Frontier Science Program (HFSP)
LinkOut - more resources
Full Text Sources
