

This is a preprint.
Human protein interactome structure prediction at scale with Boltz-2
- PMID: 40631076
- PMCID: PMC12236519
- DOI: 10.1101/2025.07.03.663068
Human protein interactome structure prediction at scale with Boltz-2
Abstract
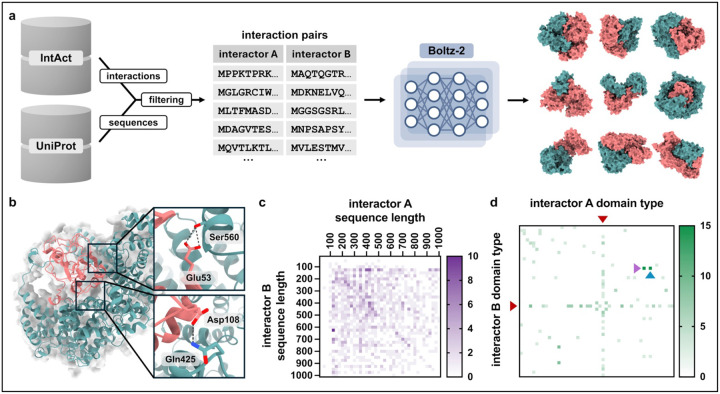
In humans, protein-protein interactions mediate numerous biological processes and are central to both normal physiology and disease. Extensive research efforts have aimed to elucidate the human protein interactome, and comprehensive databases now catalog these interactions at scale. However, structural coverage of the human protein interactome is limited and remains challenging to resolve through experimental methodology alone. Recent advances in artificial intelligence/machine learning (AI/ML)-based approaches for protein interaction structure prediction present opportunities for large-scale structural characterization of the human interactome. One such model, Boltz-2, which is capable of predicting the structures of protein complexes, may serve this objective. Here, we present de novo computed models of 1,394 binary human protein interaction structures predicted using Boltz-2 based on biochemically determined interaction data sourced from the IntAct database. We assessed the predicted interaction structures through different confidence metrics, which consider both overall structure and the interaction interface. These analyses indicated that prediction confidence tended to be greater for smaller complexes, while increased multiple sequence alignment (MSA) depth tended to improve prediction confidence. Additionally, we examined annotated protein domains and found that 679 of the predicted structural complexes contained a variety of domains with putative interaction involvement on the basis of interaction interface proximity. Furthermore, our analyses revealed intricate interaction networks within the context of biological function and cancer. This work demonstrates the utility of Boltz-2 for in silico structural modeling of the human protein interactome, highlighting both strengths and limitations, while also providing a novel view of broad functional contextualization. Ultimately, such modeling is expected to yield broad structural insights with relevance across multiple domains of biomedical research.
Conflict of interest statement
A.M.I. is a founder and partner of North Horizon, which is engaged in the development of artificial intelligence-based software. R.P. and W.A. are founders and equity shareholders of PhageNova Bio. R.P. is Chief Scientific Officer and a paid consultant of PhageNova Bio. R.P. and W.A are founders and equity shareholders of MBrace Therapeutics. R.P. and W.A. serve as paid consultants for MBrace Therapeutics. R.P. and W.A. have Sponsored Research Agreements (SRAs) in place with PhageNova Bio, MBrace Therapeutics, and Alnylam Pharmaceuticals; this study falls outside of the scope of these SRAs. These arrangements are managed in accordance with the established institutional conflict-of-interest policies of Rutgers, The State University of New Jersey. C.M. and S.K.B. declare no competing interests.
Figures





References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources