

This is a preprint.
PROFET Predicts Continuous Gene Expression Dynamics from scRNA-seq Data to Elucidate Heterogeneity of Cancer Treatment Responses
- PMID: 40631077
- PMCID: PMC12236938
- DOI: 10.1101/2025.06.27.662030
PROFET Predicts Continuous Gene Expression Dynamics from scRNA-seq Data to Elucidate Heterogeneity of Cancer Treatment Responses
Abstract
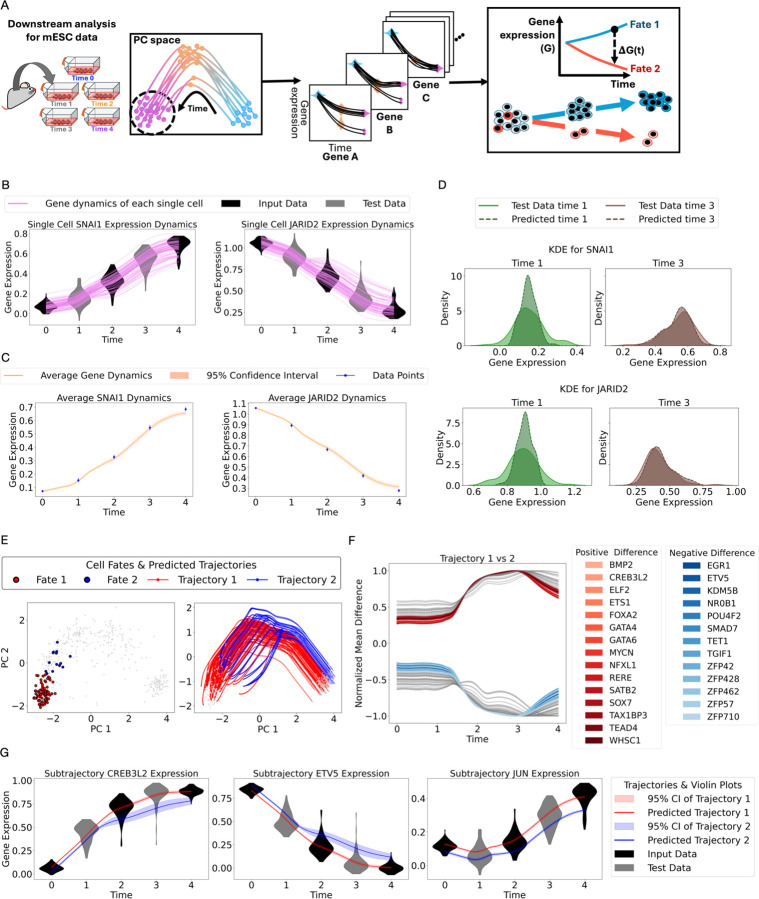
Single-cell RNA sequencing captures static snapshots of gene expression but lacks the ability to track continuous gene expression dynamics over time. To overcome this limitation, we developed PROFET (Particle-based Reconstruction Of generative Force-matched Expression Trajectories), a computational framework that reconstructs continuous, nonlinear single-cell gene expression trajectories from sparsely sampled scRNA-seq data. PROFET first generates particle flows between time-stamped samples using a novel Lipschitz-regularized gradient flow approach and then learns a global vector field for trajectory reconstruction using neural force-matching. The framework was developed using synthetic data simulating cell state transitions and subsequently validated on both mouse and human in vitro datasets. We then deployed PROFET to investigate heterogeneity in treatment responses to palbociclib, a CDK4/6 inhibitor, in hormone receptor positive breast cancer. By comparing newly generated scRNA-seq data from a palbociclib-resistant breast cancer cell line with published patient-derived datasets, we identified a subpopulation of patient cells exhibiting profound phenotypic shifts in response to treatment, along with surface markers uniquely enriched in those cells. By recovering temporal information from static snapshots, PROFET enables inference of continuous single-cell expression trajectories, providing a powerful tool for dissecting the heterogeneity of cell state transitions in treatment responses.
Figures





References
-
- Skylaki S., Hilsenbeck O. & Schroeder T. Challenges in long-term imaging and quantification of single-cell dynamics. Nat Biotechnol 34, 1137–1144 (2016). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources