

This is a preprint.
Product-stabilized filamentation by human glutamine synthetase allosterically tunes metabolic activity
- PMID: 40631248
- PMCID: PMC12236511
- DOI: 10.1101/2025.07.04.663231
Product-stabilized filamentation by human glutamine synthetase allosterically tunes metabolic activity
Abstract
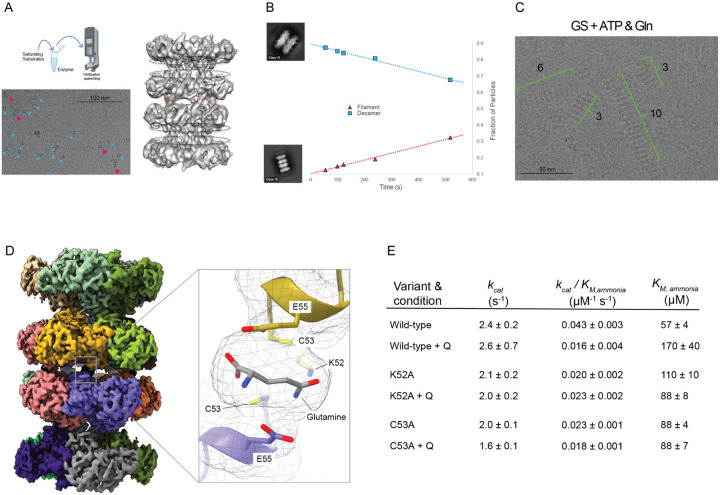
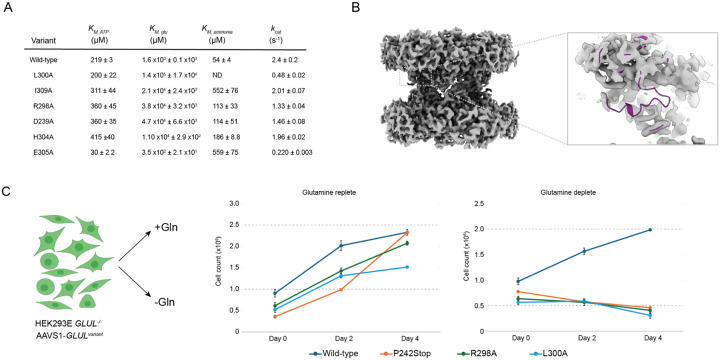
To maintain metabolic homeostasis, enzymes must adapt to fluctuating nutrient levels through mechanisms beyond gene expression. Here, we demonstrate that human glutamine synthetase (GS) can reversibly polymerize into filaments aided by a composite binding site formed at the filament interface by the product, glutamine. Time-resolved cryo-electron microscopy (cryo-EM) confirms that glutamine binding stabilizes these filaments, which in turn exhibit reduced catalytic specificity for ammonia at physiological concentrations. This inhibition appears induced by a conformational change that remodulates the active site loop ensemble gating substrate entry. Metadynamics ensemble refinement revealed >10 Å conformational range for the active site loop and that the loop is stabilized by transient contacts. This disorder is significant, as we show that the transient contacts which stabilize this loop in a closed conformation are essential for catalysis both in vitro and in cells. We propose that GS filament formation constitutes a negative-feedback mechanism, directly linking product concentration to the structural and functional remodeling of the enzyme.
Conflict of interest statement
Conflicts of interest HY is an employee of JSR. J.S.F. is a consultant to, a shareholder of, and receives sponsored research support from Relay Therapeutics.
Figures




References
-
- Abraham Mark James, Murtola Teemu, Schulz Roland, Páll Szilárd, Smith Jeremy C., Hess Berk, and Lindahl Erik. 2015. “GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers.” SoftwareX 1–2 (September):19–25.
-
- Albe K. R., Butler M. H., and Wright B. E.. 1990. “Cellular Concentrations of Enzymes and Their Substrates.” Journal of Theoretical Biology 143 (2): 163–95. - PubMed
-
- Arad G., Freikopf A., and Kulka R. G.. 1976. “Glutamine-Stimulated Modification and Degradation of Glutamine Synthetase in Hepatoma Tissue Culture Cells.” Cell 8 (1): 95–101. - PubMed
-
- Atkinson D. E. 1965. “Biological Feedback Control at the Molecular Level.” Science (New York, N.Y.) 150 (3698): 851–57. - PubMed
-
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., and Haak J. R.. 1984. “Molecular Dynamics with Coupling to an External Bath.” The Journal of Chemical Physics 81 (8): 3684–90.