

Integrated Nanopore and short-read RNA sequencing identifies dysregulation of METTL3- m6A modifications in endocrine therapy- sensitive and resistant breast cancer cells
- PMID: 40632306
- PMCID: PMC12241209
- DOI: 10.1007/s10142-025-01658-2
Integrated Nanopore and short-read RNA sequencing identifies dysregulation of METTL3- m6A modifications in endocrine therapy- sensitive and resistant breast cancer cells
Abstract
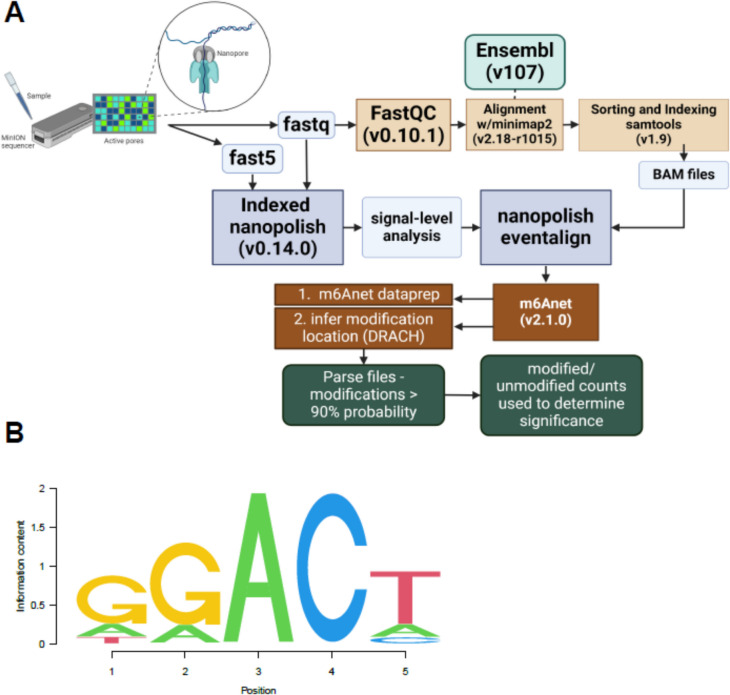
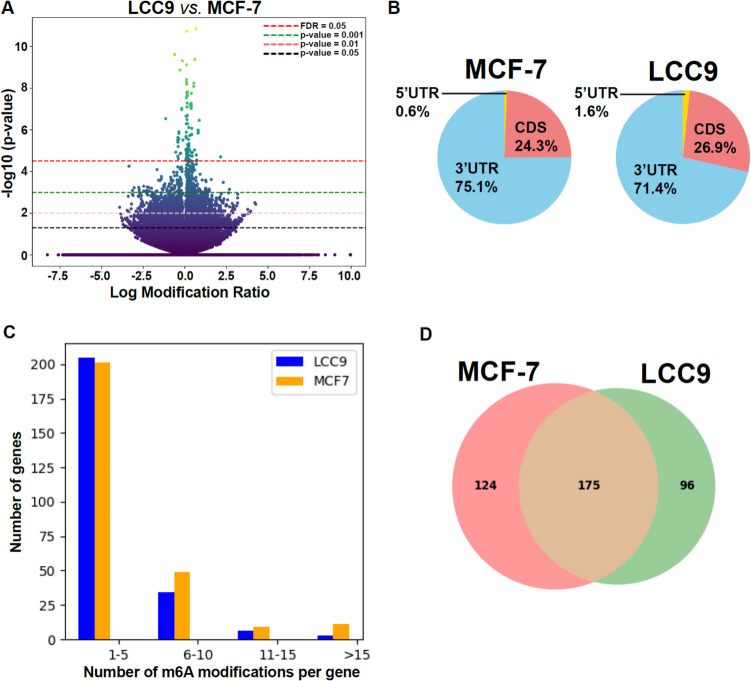
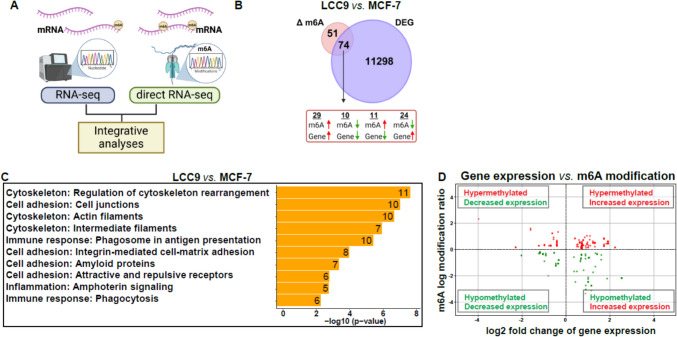
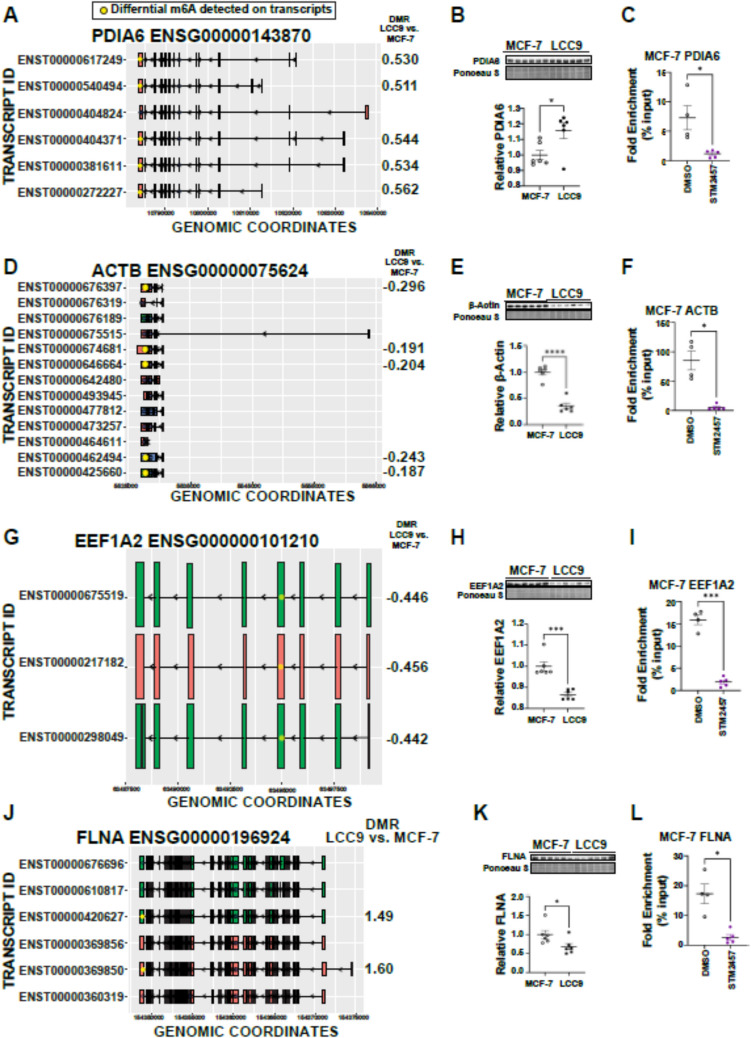
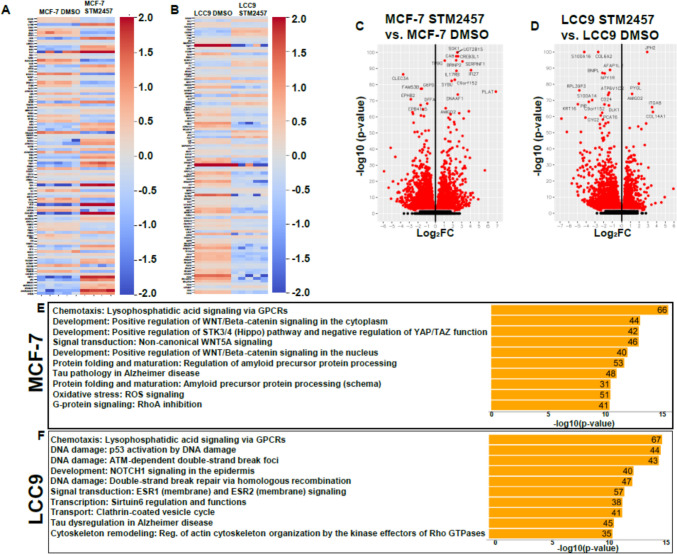
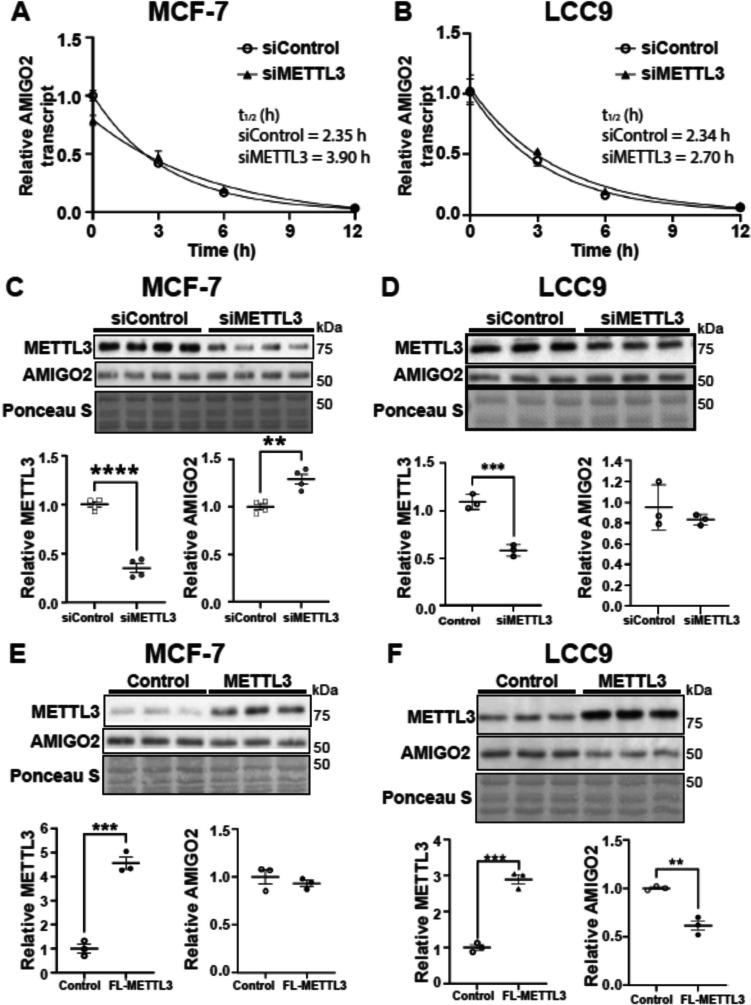
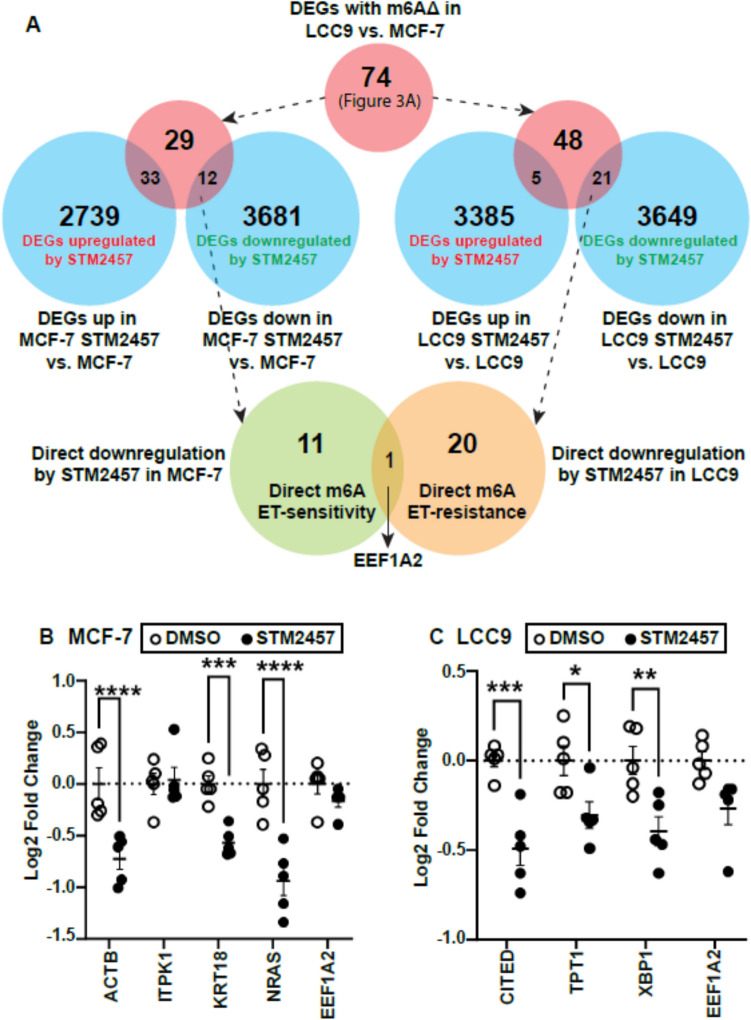
The role of epitranscriptomic changes in the development of acquired endocrine therapy (ET)- resistance in estrogen receptor α (ER) expressing breast cancer (BC) is unknown. We tested the hypothesis that inhibition of METTL3, the methyltransferase responsible for the mRNA modification N-6 methyladenosine (m6A), alters m6A modifications and differentially regulates the abundance of mRNA transcripts in ET-sensitive MCF-7 versus resistant LCC9 ER + human BC cells. Differential m6A modifications were identified using direct mRNA sequencing (DRS) performed on five replicates for each cell line ± 1 µM STM2457, a selective METTL3 inhibitor, using Nanopore MinION long read RNA-seq. Parallel short read Illumina RNA-seq quantified differential transcript abundance in the same samples. Selected results were validated by RT-qPCR, m6A-RIP-qPCR, reporter assays, and western blot analysis. Statistical analysis combined m6Anet, a machine-learning algorithm designed to call m6A modified bases, with a generalized linear model following a binomial distribution analysis to identify significant differential m6A modification ratios (DMR). Distinct METTL3 dependent m6A modification patterns in LCC9 and MCF-7 cells were observed in differentially expressed genes (DEG) associated with ET-resistance, including EEF1A2, ACTB, FLNA, PDIA6, AMIGO2, TPT1, XBP1, and CITED4. Select results were validated in additional ET-resistant BC cell lines. m6A-RIP-RT-qPCR validated specific m6A sites. We examined the proximity of m6A sites to estrogen receptor α (ER α)-mRNA binding sites reported in MCF-7 cells. ACTB, PDIA6, and XBP1 demonstrated a short-range proximity, with m6A sites located within 100 bp of ERα binding sites, suggesting a role for m6A in influencing ERα-mRNA binding. Our work provides a framework for integrating DRS and DEG omics data. Our results suggest a role for dysregulation of m6A modifications in pathways implicated in ET resistance in BC.
Keywords: Breast cancer; Endocrine-resistance; Epitranscriptome; M6A; Nanopore DRS.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Conflict of interest: The authors declare no competing interests. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
PARP-1 as a novel target in endocrine-resistant breast cancer.J Exp Clin Cancer Res. 2025 Jun 16;44(1):175. doi: 10.1186/s13046-025-03441-4. J Exp Clin Cancer Res. 2025. PMID: 40518539 Free PMC article.
-
Exploring the m5C epitranscriptome of mRNAs in breast cancer cells through genome engineering and long-read sequencing approaches.Funct Integr Genomics. 2025 Jun 25;25(1):136. doi: 10.1007/s10142-025-01648-4. Funct Integr Genomics. 2025. PMID: 40555775 Free PMC article.
-
Alteration of m6A Methylation in Breast Cancer Cells by Kalanchoe pinnata Aqueous Extract.Molecules. 2025 Jun 18;30(12):2634. doi: 10.3390/molecules30122634. Molecules. 2025. PMID: 40572597 Free PMC article.
-
RNA modifications in female reproductive physiology and disease: emerging roles and clinical implications.Hum Reprod Update. 2025 Jul 1;31(4):333-360. doi: 10.1093/humupd/dmaf005. Hum Reprod Update. 2025. PMID: 40152541 Review.
-
RNA N 6-Methyladenosine Modification in Normal and Malignant Hematopoiesis.Adv Exp Med Biol. 2019;1143:75-93. doi: 10.1007/978-981-13-7342-8_4. Adv Exp Med Biol. 2019. PMID: 31338816 Review.
References
-
- Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, Ramos P, Tapia C, Kim HR, Yerrum S, Sharma CG et al (2011) Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A 108(47):18879–18886 - PMC - PubMed
-
- Bartha Á, Győrffy B (2021) TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int J Mol Sci. 22(5) - PMC - PubMed
-
- Begik O, Lucas MC, Pryszcz LP, Ramirez JM, Medina R, Milenkovic I, Cruciani S, Liu H, Vieira HGS, Sas-Chen A et al (2021) Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat Biotechnol 39(10):1278–1291 - PubMed
MeSH terms
Substances
Grants and funding
- HT9425-23-1-0017/Office of the Assistant Secretary of Defense for Health Affairs Breast Cancer Research Program
- 5P20GM103436-22/National Institutes of Health National Institute of General Medical Sciences
- T35 DK072923/DK/NIDDK NIH HHS/United States
- T35 DK072923/NH/NIH HHS/United States
- P20 GM103436/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
