

Decomposition of phenotypic heterogeneity in autism reveals underlying genetic programs
- PMID: 40634707
- PMCID: PMC12283356
- DOI: 10.1038/s41588-025-02224-z
Decomposition of phenotypic heterogeneity in autism reveals underlying genetic programs
Abstract
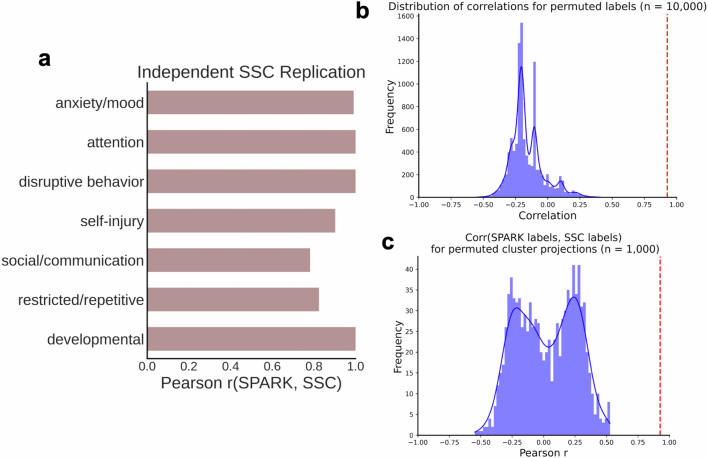
Unraveling the phenotypic and genetic complexity of autism is extremely challenging yet critical for understanding the biology, inheritance, trajectory and clinical manifestations of the many forms of the condition. Using a generative mixture modeling approach, we leverage broad phenotypic data from a large cohort with matched genetics to identify robust, clinically relevant classes of autism and their patterns of core, associated and co-occurring traits, which we further validate and replicate in an independent cohort. We demonstrate that phenotypic and clinical outcomes correspond to genetic and molecular programs of common, de novo and inherited variation and further characterize distinct pathways disrupted by the sets of mutations in each class. Remarkably, we discover that class-specific differences in the developmental timing of affected genes align with clinical outcome differences. These analyses demonstrate the phenotypic complexity of children with autism, identify genetic programs underlying their heterogeneity, and suggest specific biological dysregulation patterns and mechanistic hypotheses.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













Update of
-
Decomposition of phenotypic heterogeneity in autism reveals distinct and coherent genetic programs.medRxiv [Preprint]. 2024 Aug 16:2024.08.15.24312078. doi: 10.1101/2024.08.15.24312078. medRxiv. 2024. Update in: Nat Genet. 2025 Jul;57(7):1611-1619. doi: 10.1038/s41588-025-02224-z. PMID: 39185525 Free PMC article. Updated. Preprint.
References
-
- Diagnostic and Statistical Manual of Mental Disorders 5th edn (American Psychiatric Association, 2013).
-
- Simonoff, E. et al. Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry47, 921–929 (2008). - PubMed
MeSH terms
Grants and funding
- T32HG003284/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01GM071966/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01 GM071966/GM/NIGMS NIH HHS/United States
- 395506/Simons Foundation
- T32 HG003284/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
