

Using Deep Graph Neural Networks Improves Physics-Based Hydration Free Energy Predictions Even for Molecules Outside of the Training Set Distribution
- PMID: 40641374
- PMCID: PMC12302205
- DOI: 10.1021/acs.jpcb.5c02263
Using Deep Graph Neural Networks Improves Physics-Based Hydration Free Energy Predictions Even for Molecules Outside of the Training Set Distribution
Abstract
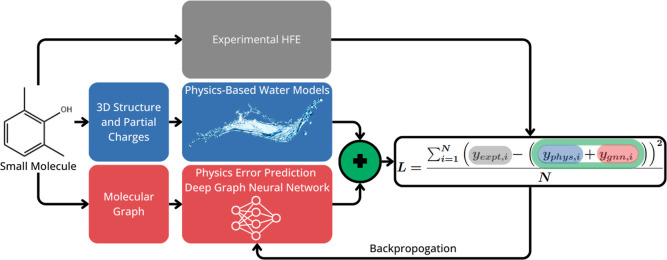
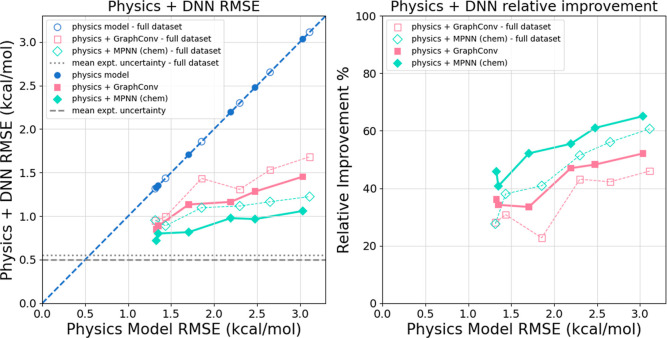
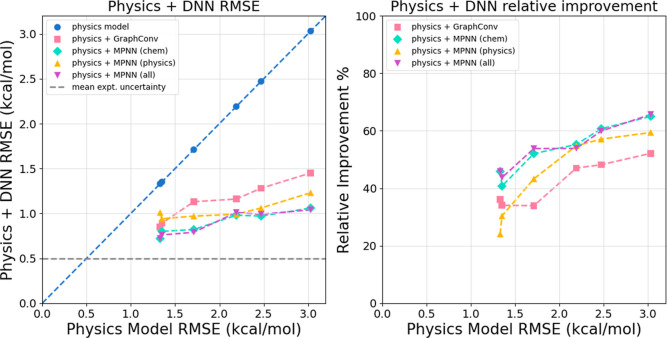
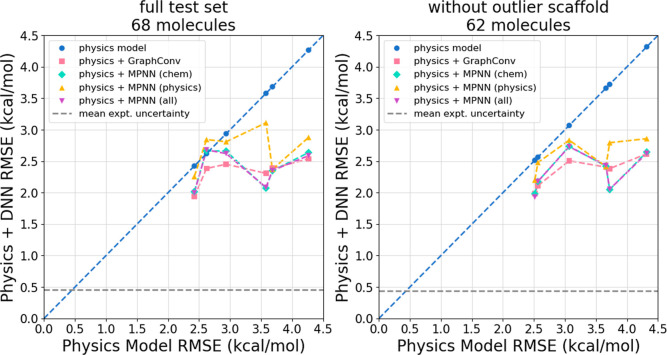
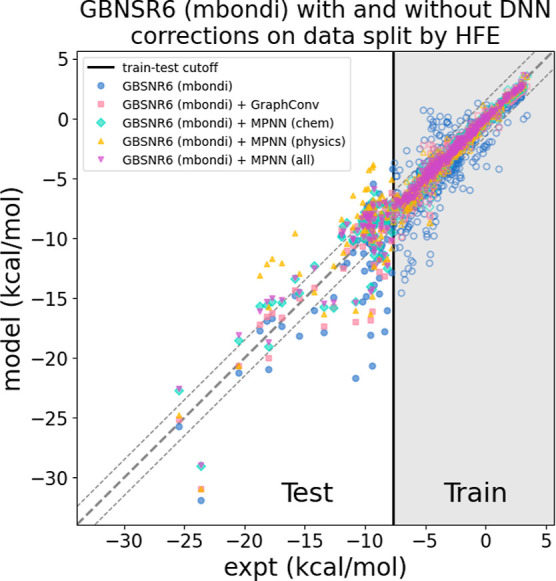
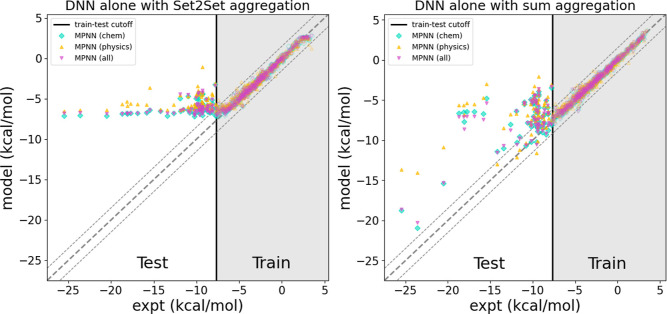
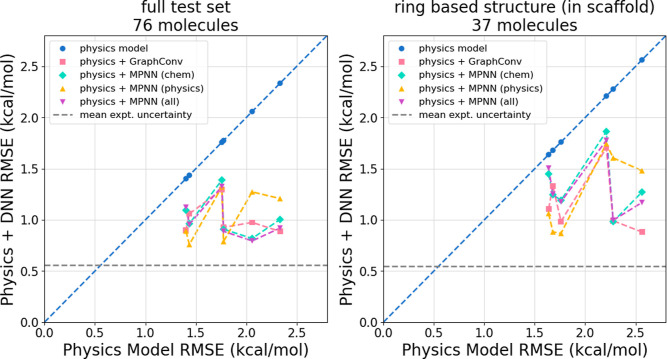
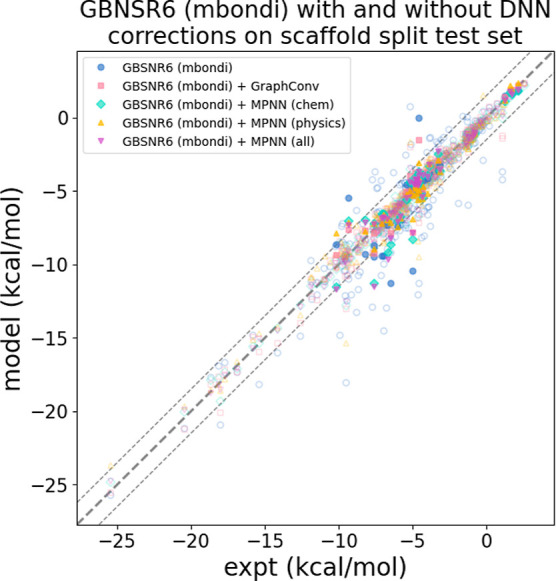
The accuracy of computational water models is crucial to atomistic simulations of biomolecules. Here we explore a decoupled framework that combines classical physics-based models with deep neural networks (DNNs) to correct residual error in hydration free energy (HFE) prediction. Our main goal is to evaluate this framework on out-of-distribution data (molecules that differ significantly from those used in training), where DNNs are known to struggle. Several common physics-based solvation models are used in the evaluation. Graph neural network architectures are tested for their ability to generalize using multiple data set splits, including out-of-distribution HFEs and unseen molecular scaffolds. Our most important finding is that for out-of-distribution data, where DNNs alone often struggle, the physics + DNN models consistently improve physics model predictions. For in-distribution data, the DNN corrections significantly improve the accuracy of physics-based models, with a final RMSE below 1 kcal/mol and a relative improvement between 40% and 65% in most cases. The accuracy of physics + DNN models tends to improve when the 6% of molecules with the highest experimental uncertainty are removed. This study provides insights into the potential and limitations of combining physics and machine learning for molecular modeling, offering a practical and generalizable strategy of using DNN as independent postprocessing correction.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
