

Apolipoprotein L1 (APOL1): Consideration of Molecular Evolution, Interaction with APOL3, and Impact of Splice Isoforms Advances Understanding of Cellular and Molecular Mechanisms of Cell Injury
- PMID: 40643530
- PMCID: PMC12248677
- DOI: 10.3390/cells14131011
Apolipoprotein L1 (APOL1): Consideration of Molecular Evolution, Interaction with APOL3, and Impact of Splice Isoforms Advances Understanding of Cellular and Molecular Mechanisms of Cell Injury
Abstract
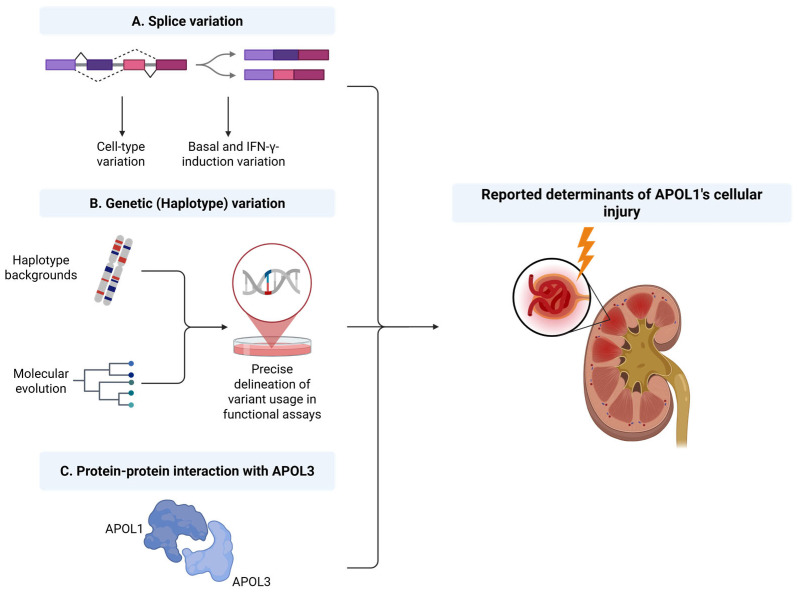
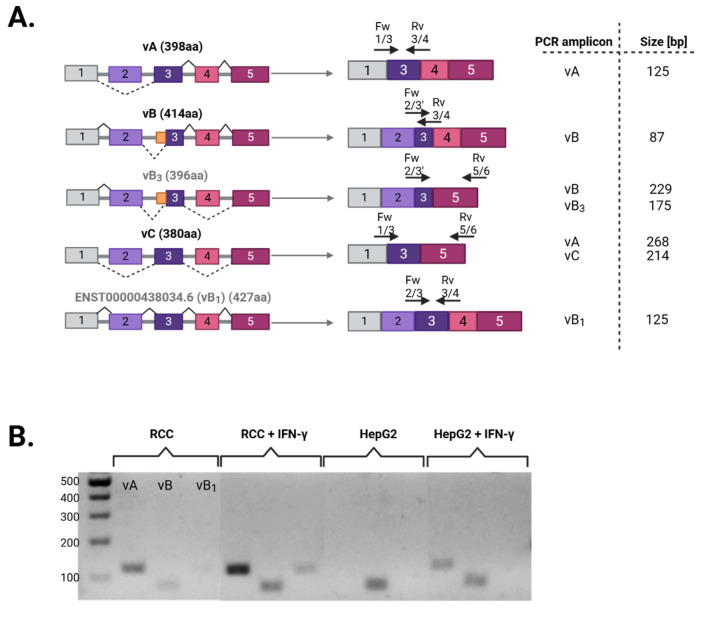
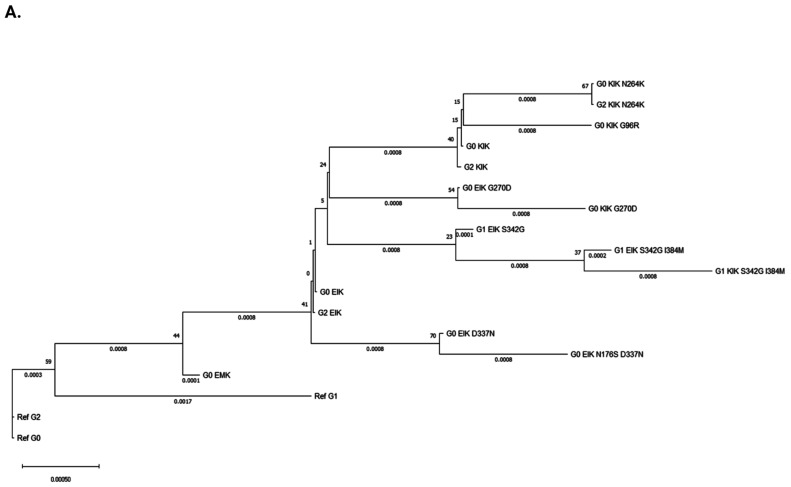
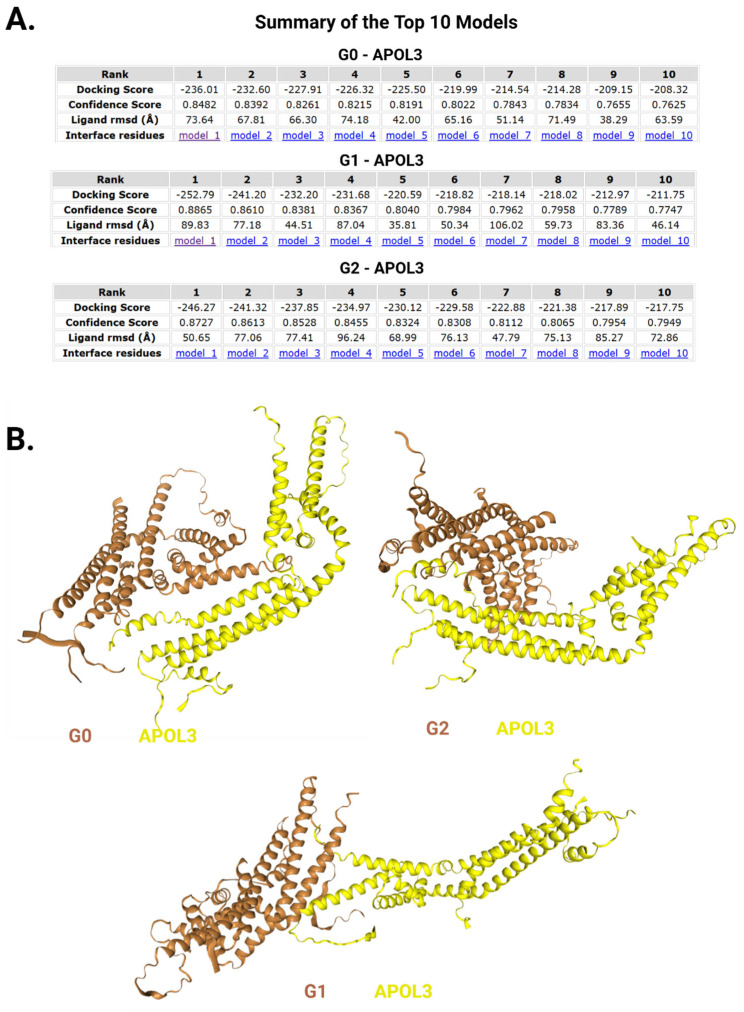
The Apolipoprotein L1 (APOL1) innate immunity gene product represents the sole member of the APOL gene family in humans capable of secretion into circulation, thereby mediating the trypanolysis of T. brucei brucei. Gain-of-function variants of the APOL1 gene originated and spread among human population groups to extend APOL1's protective capacity to include also serum-resistant subspecies, such as T. brucei gambiense (S342G known as APOL1-G1) and T. brucei rhodesiense (N388_Y389del known as APOL1-G2). The biochemical pathways underlying the lytic activity of these evolutionary favored mutations against bloodstream trypanosomes have been elucidated with remarkable precision. However, the intricate molecular mechanisms by which such variants confer an increased susceptibility to renal cellular injury and consequent kidney disease remain incompletely defined. In the absence of a consistent mechanistic explanation for differential kidney injury, we propose pursuing three interrelated avenues of investigation informed by prior epidemiological and mechanistic evidence linking them to APOL1's cytotoxicity: (1) Molecular evolution of APOL1 haplotypes in human populations, (2) APOL1 splicing and consequent splice isoforms, (3) Interaction of APOL1 with other APOL gene family members, prioritizing APOL3. In the current study, we use reanalysis of population genetics datasets to resolve the haplotype contexts of all protein-altering APOL1 variants, uncovering previously unrecognized variant-haplotype couplings. We further characterize distinct cellular physiological properties among APOL1 splice isoforms, stressing the importance of isoform vB and what can be learned from isoform vC. Finally, a native interaction, and its interface, between APOL1 and APOL3 is reported, and shown to be differentially modulated by G1 and G2. We contend that continuing studies integrating these three interrelated domains will substantially advance mechanistic insights into APOL1 variant-driven renal injury, and leverage the findings to provide a more cohesive framework to guide future research.
Keywords: APOL1 risk variants; APOL1 splice variants; APOL3; phylogenetics; protein–protein interaction; signal peptide; trypanosome lytic factor; trypanosomiasis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures








References
-
- Stephens J.W.W., Fantham H.B. On the peculiar morphology of a trypanosome from a case of sleeping sickness and the possibility of its being a new species (T. rhodesiense) Proc. R. Soc. Lond. Ser. B Contain. Pap. A Biol. Character. 1910;83:28–33. doi: 10.1080/00034983.1910.11685723. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
