

Structural and Functional Characterization of N-Glycanase-1 Pathogenic Variants
- PMID: 40643555
- PMCID: PMC12248763
- DOI: 10.3390/cells14131036
Structural and Functional Characterization of N-Glycanase-1 Pathogenic Variants
Abstract
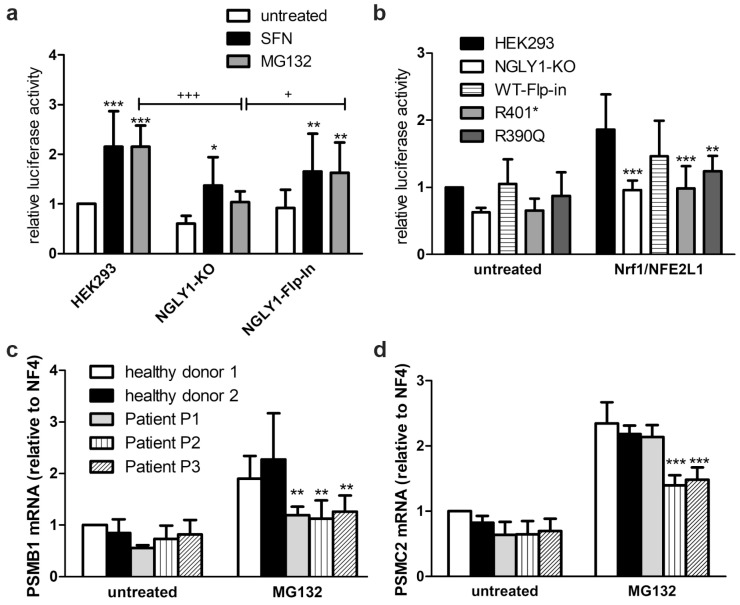
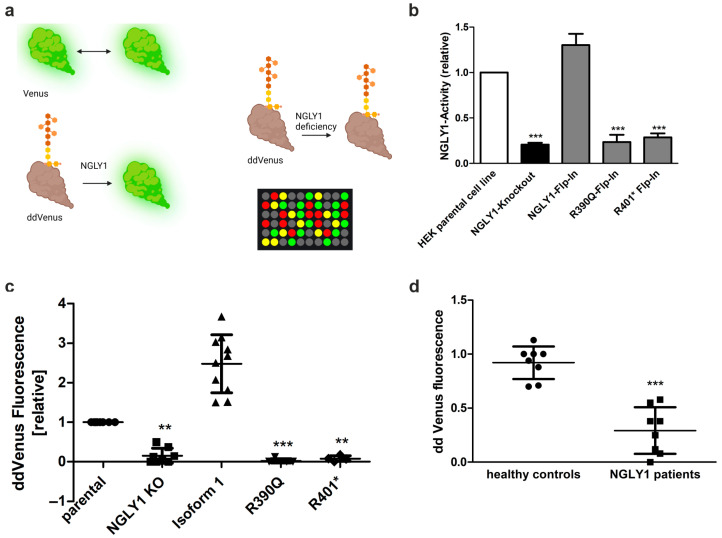
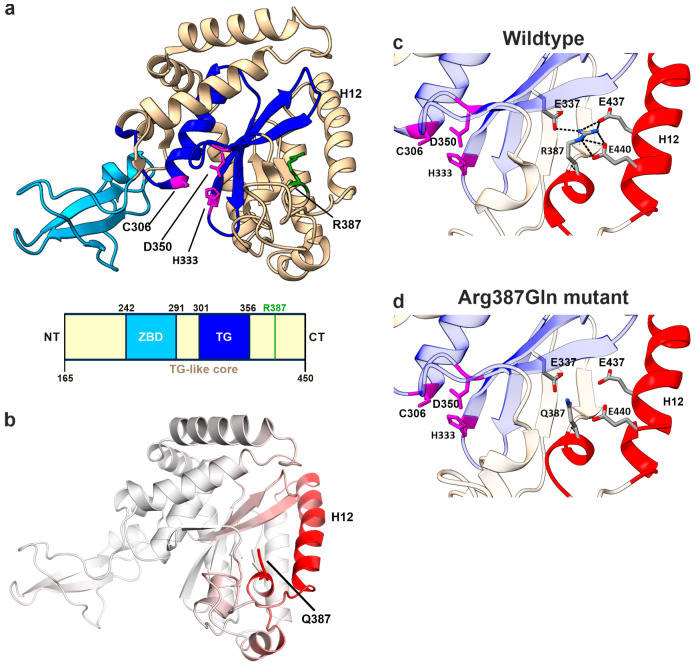
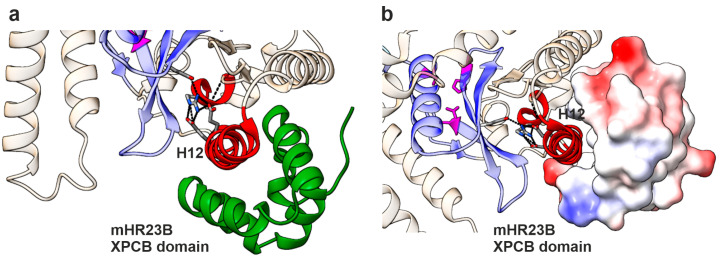
NGLY1 deficiency is a congenital disorder of deglycosylation, caused by pathogenic variants of the NGLY1 gene. It manifests as global developmental delay, hypo- or alacrima, hypotonia, and a primarily hyperkinetic movement disorder. The NGLY1 enzyme is involved in deglycosylation of misfolded N-glycosylated proteins before their proteasomal degradation and in the activation of transcription factors that control the expression of proteasomal subunits. Here, we have characterized the pathogenic NGLY1 variants found in three Swiss NGLY deficiency patients, as well as the most common pathogenic NGLY1 variant, Arg401*, found in about 20% of patients. Our functional and structural assessments of these variants show that they cause a profound reduction in NGLY1 activity, severely reduced expression of NGLY1 protein, and misprocessing of the transcription factor NFE2L1. Furthermore, transcription of proteasomal subunits and NGLY1 mRNA splicing are impaired by some of these variants. Our in silico structural analysis shows that the Arg390Gln substitution results in destabilization of NGLY1 structure due to a loss of an ionic interaction network of Arg390 and potentially impairment of protein-protein interactions. Our results provide important information on the functional and structural effects of pathogenic NGLY1 variants and pave the way for structure-based development of personalized treatment options.
Keywords: ERAD; N-Glycosylation; congenital disorders of deglycosylation; developmental delay; proteasome; protein misfolding.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Huang C., Harada Y., Hosomi A., Masahara-Negishi Y., Seino J., Fujihira H., Funakoshi Y., Suzuki T., Dohmae N., Suzuki T. Endo-β-N-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. USA. 2015;112:1398–1403. doi: 10.1073/pnas.1414593112. - DOI - PMC - PubMed
-
- Zhao G., Zhou X., Wang L., Li G., Kisker C., Lennarz W.J., Schindelin H. Structure of the mouse peptide N-glycanase-HR23 complex suggests co-evolution of the endoplasmic reticulum-associated degradation and DNA repair pathways. J. Biol. Chem. 2006;281:13751–13761. doi: 10.1074/jbc.M600137200. - DOI - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
