

RIT1 Drives Oncogenic Transformation and is an Actionable Target in Lung Adenocarcinoma
- PMID: 40644578
- PMCID: PMC12281470
- DOI: 10.1158/0008-5472.CAN-24-3819
RIT1 Drives Oncogenic Transformation and is an Actionable Target in Lung Adenocarcinoma
Abstract
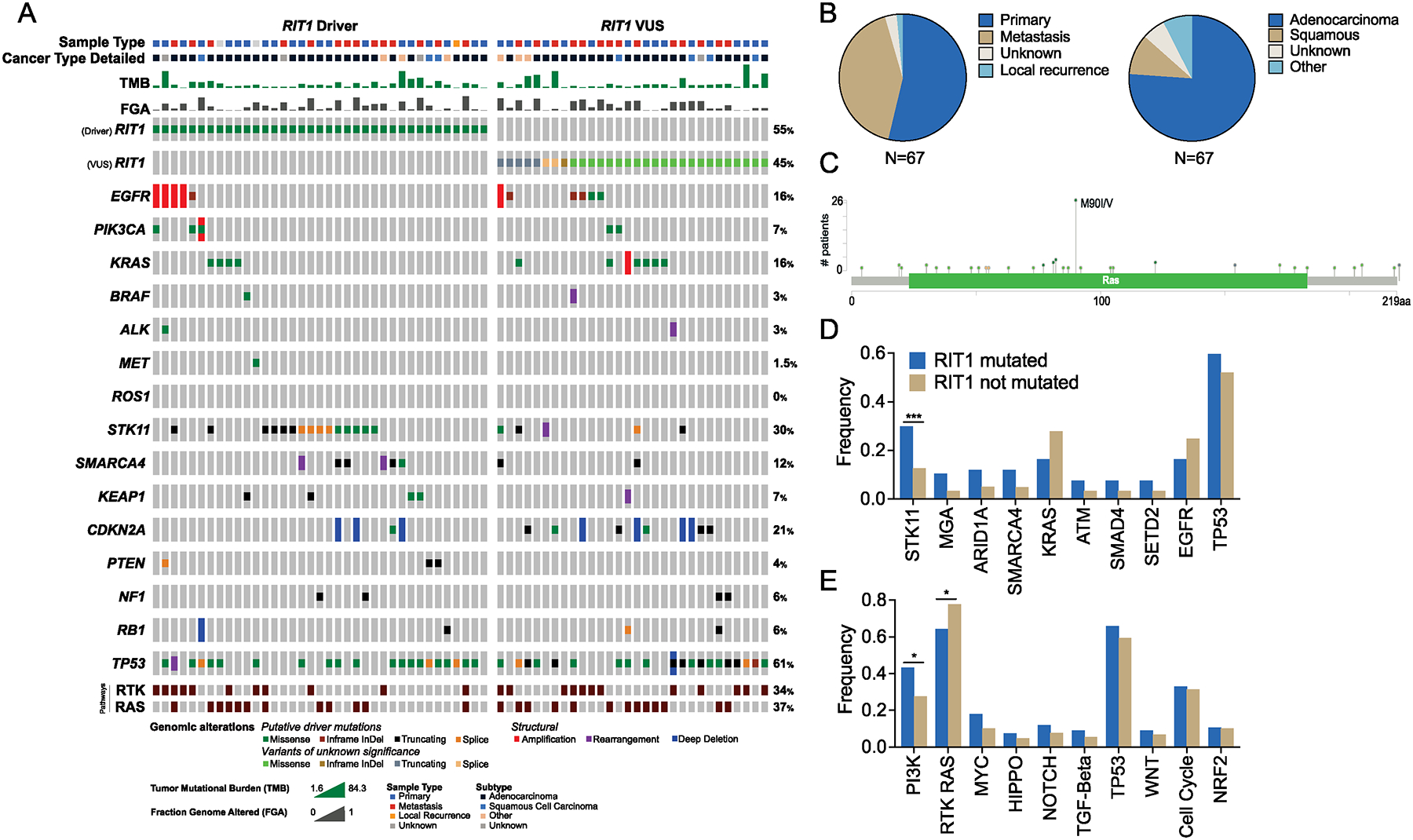
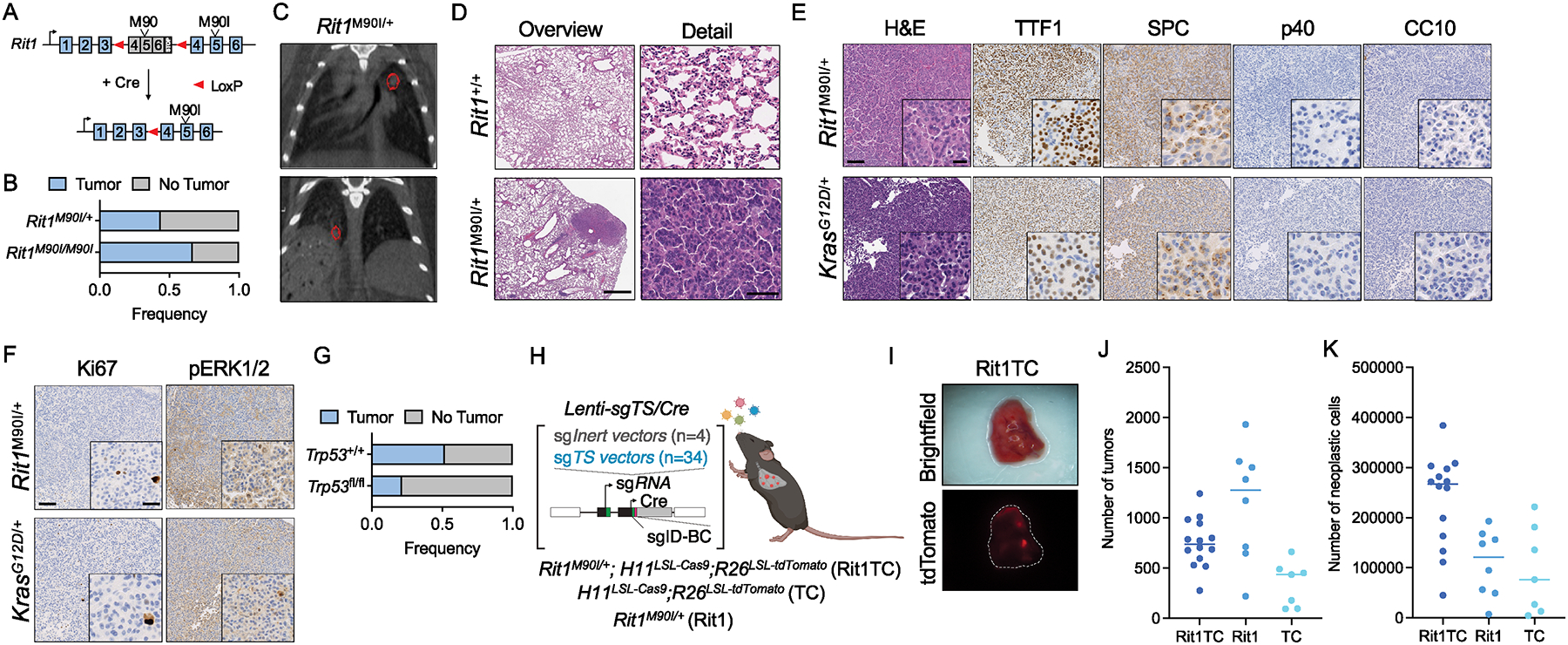
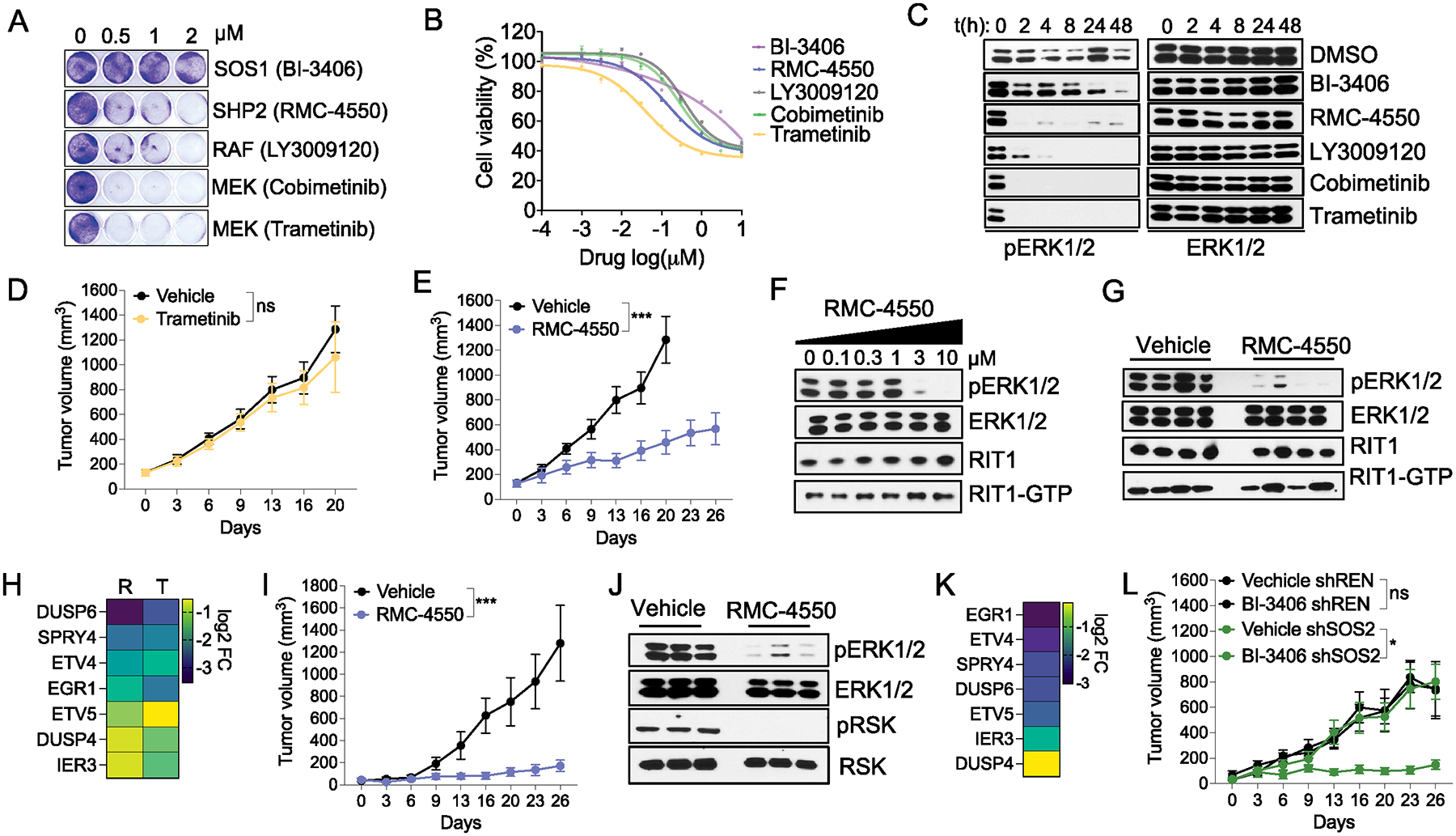
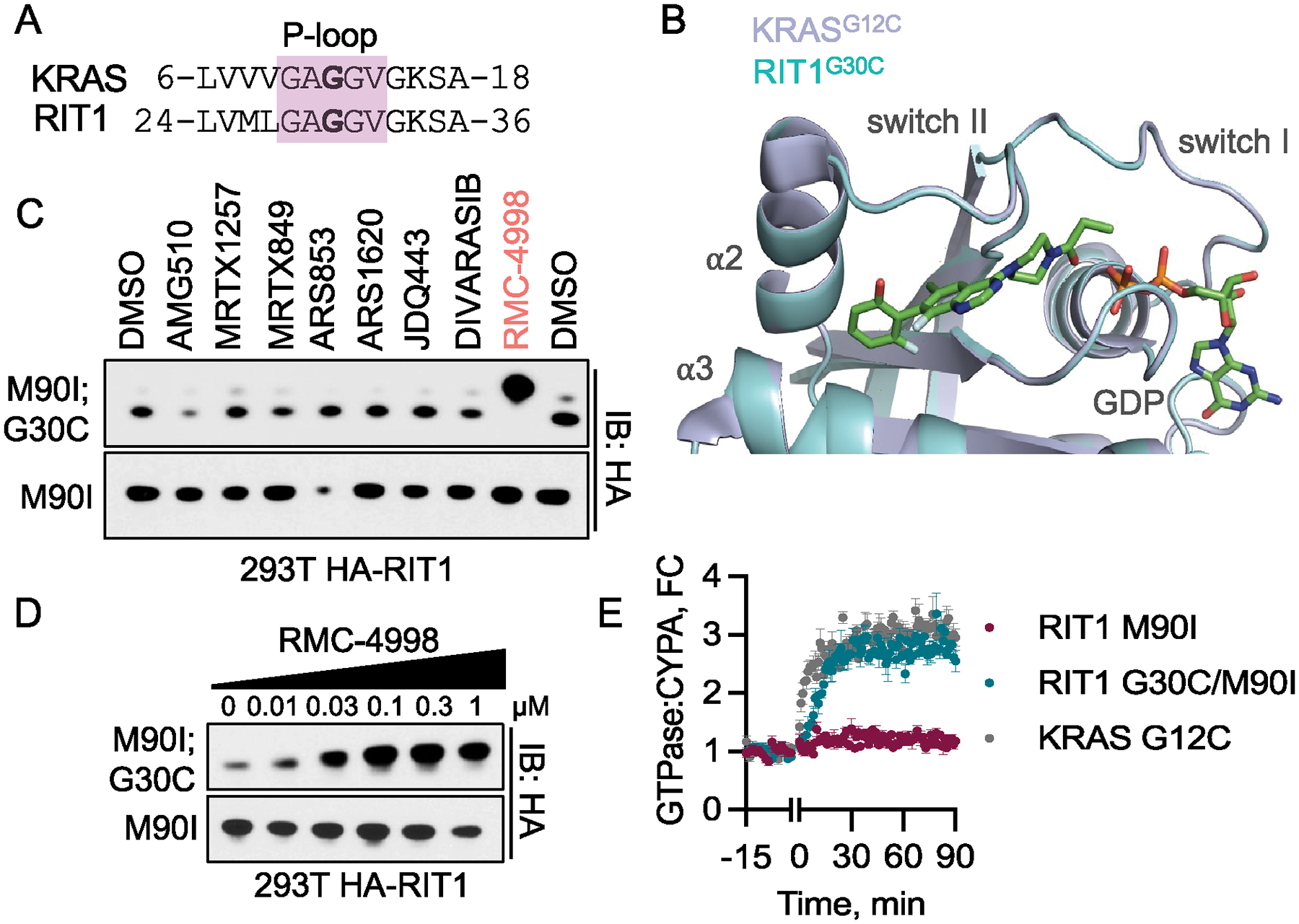
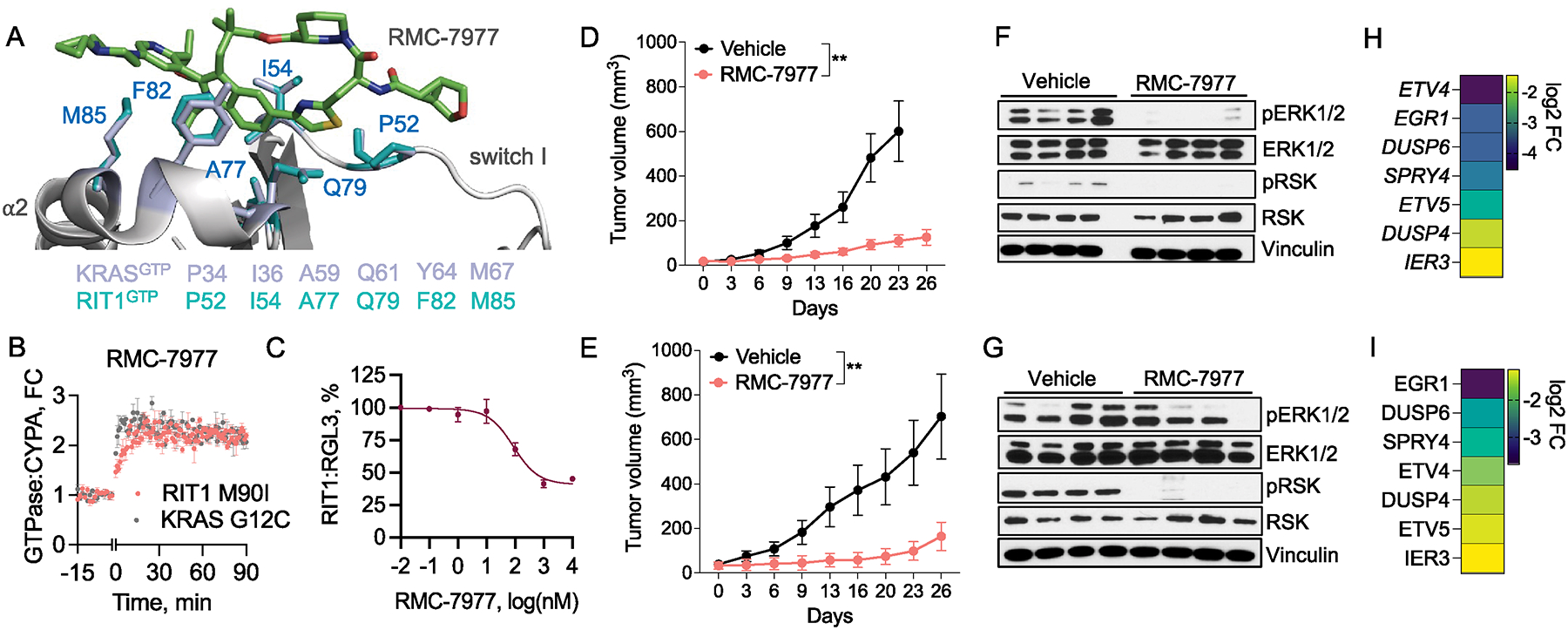
RIT1 is a small GTPase of the RAS family, and RIT1 mutations have been identified in lung cancer, leukemias, and the developmental disorder Noonan syndrome. Mutations in RIT1 lead to increased protein levels due to impaired proteolysis, resulting in dysregulation of RAS/MAPK signaling and other pathways. Here, we documented the diversity of RIT1 mutations in human lung cancer and showed that physiological expression of RIT1 M90I is sufficient to drive autochthonous lung tumor development in vivo in mouse models. Evaluation of complementary methods to either inhibit RIT1 directly or the downstream RAS/MAPK pathway revealed that RIT1 M90I tumors are sensitive to SHP2 inhibitors and RAS nucleotide exchange inhibition. Additionally, a proof-of-concept chemical biology approach identified that RAS tri-complex inhibitors bind directly to GTP-bound RIT1, resulting in tumor shrinkage. These molecules provide a feasible therapeutic approach for RIT1-driven lung tumors.
Conflict of interest statement
Conflict of interest statement:
The authors declare no potential conflicts of interest related to this work.
Figures
Similar articles
-
RIT1M90I is a driver of lung adenocarcinoma tumorigenesis and resistance to targeted therapy.Cancer Res. 2025 Jul 3. doi: 10.1158/0008-5472.CAN-24-3662. Online ahead of print. Cancer Res. 2025. PMID: 40608319
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Targeting the SHOC2-RAS interaction in RAS-mutant cancers.Nature. 2025 Jun;642(8066):232-241. doi: 10.1038/s41586-025-08931-1. Epub 2025 May 7. Nature. 2025. PMID: 40335703 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2020 Jan 9;1(1):CD011535. doi: 10.1002/14651858.CD011535.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2021 Apr 19;4:CD011535. doi: 10.1002/14651858.CD011535.pub4. PMID: 31917873 Free PMC article. Updated.
References
Grants and funding
LinkOut - more resources
Full Text Sources
