

Privacy-preserving multicenter differential protein abundance analysis with FedProt
- PMID: 40646319
- PMCID: PMC12374843
- DOI: 10.1038/s43588-025-00832-7
Privacy-preserving multicenter differential protein abundance analysis with FedProt
Abstract
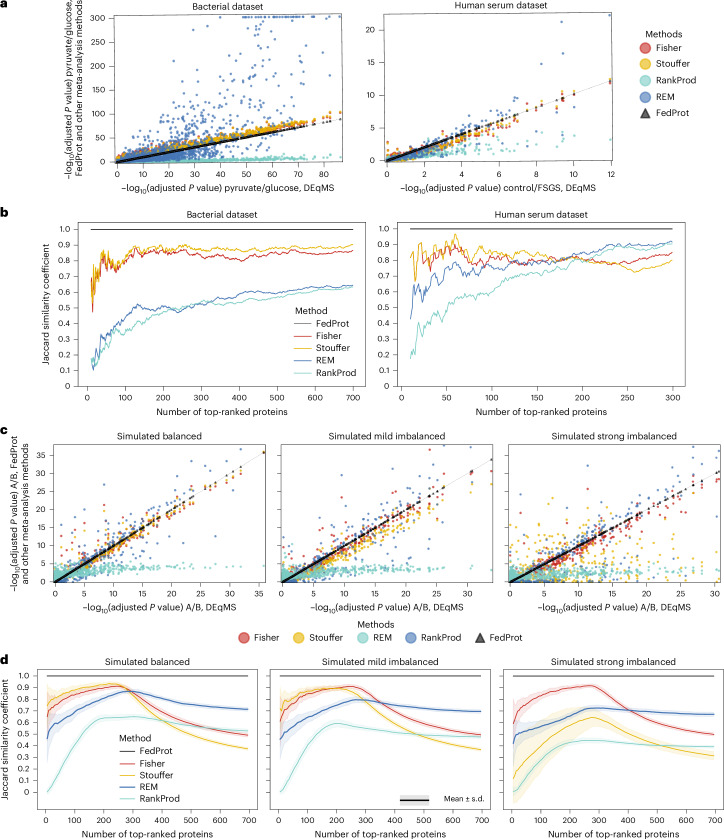
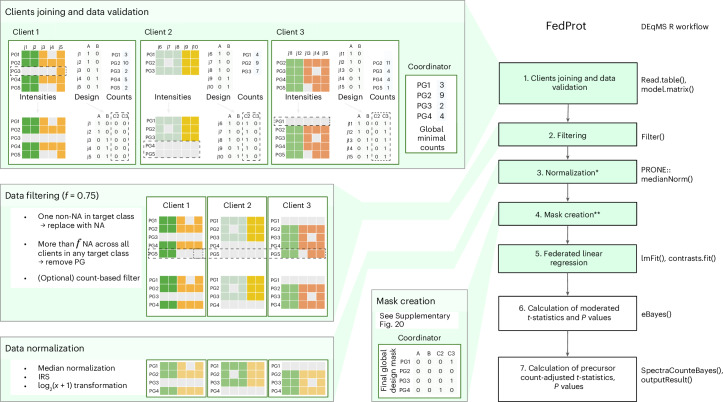
Quantitative mass spectrometry has revolutionized proteomics by enabling simultaneous quantification of thousands of proteins. Pooling patient-derived data from multiple institutions enhances statistical power but raises serious privacy concerns. Here we introduce FedProt, the first privacy-preserving tool for collaborative differential protein abundance analysis of distributed data, which utilizes federated learning and additive secret sharing. In the absence of a multicenter patient-derived dataset for evaluation, we created two: one at five centers from E. coli experiments and one at three centers from human serum. Evaluations using these datasets confirm that FedProt achieves accuracy equivalent to the DEqMS method applied to pooled data, with completely negligible absolute differences no greater than 4 × 10-12. By contrast, -log10P computed by the most accurate meta-analysis methods diverged from the centralized analysis results by up to 25-26.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: B.K. is a co-founder and shareholder of MSAID; he holds no operational role in the company. The other authors declare no competing interests.
Figures

References
-
- Aebersold, R. & Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature537, 347–355 (2016). - PubMed
-
- Altelaar, A. F. M., Munoz, J. & Heck, A. J. R. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet.14, 35–48 (2013). - PubMed
-
- Muntel, J. et al. Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC–MS instrumentation and data analysis strategy. Mol. Omics15, 348–360 (2019). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
