

A novel β-TrCP1/NRF2 interaction inhibitor for effective anti-inflammatory therapy
- PMID: 40646550
- PMCID: PMC12247323
- DOI: 10.1186/s12929-025-01157-3
A novel β-TrCP1/NRF2 interaction inhibitor for effective anti-inflammatory therapy
Abstract
Background: Non-communicable chronic diseases are characterized by low-grade inflammation and oxidative stress. Extensive research has identified the transcription factor NRF2 as a potential therapeutic target. Current NRF2 activators, designed to inhibit its repressor KEAP1, often exhibit undesirable side effects. As an alternative approach, we previously developed PHAR, a protein-protein interaction inhibitor of β-TrCP1/NRF2, which promotes NRF2 activation. Using the same in silico screening platform, we have now identified a novel compound, P10. This small molecule selectively interferes with the β-TrCP1/NRF2 interaction, leading to NRF2 stabilization and transcriptional activation of its target genes in a β-TrCP1-dependent manner, demonstrating promising effects in a liver model of acute inflammation.
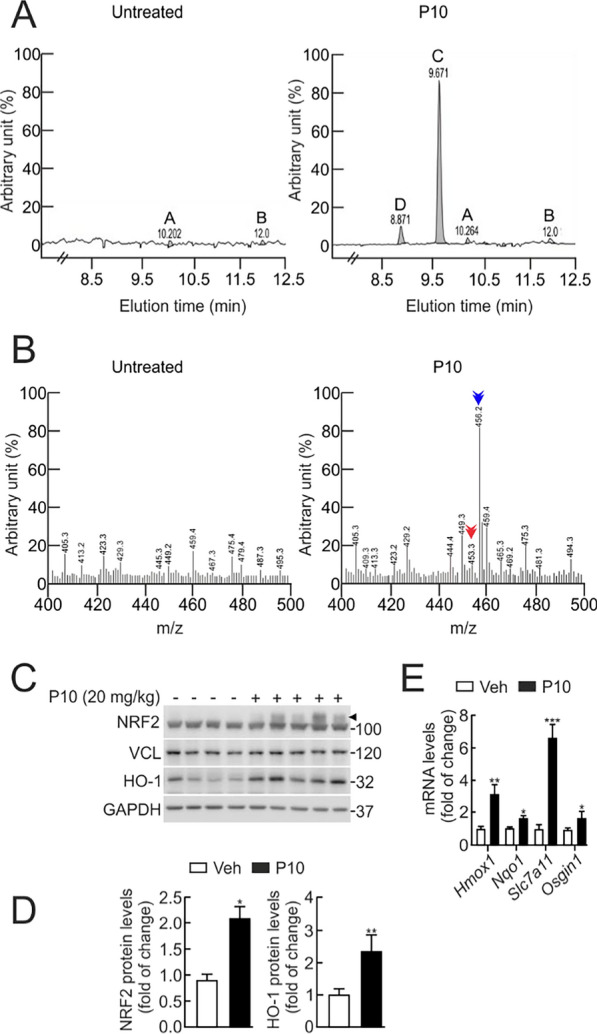
Methods: After an in silico screening of ∼1 million compounds, including molecular docking analysis, ADMET evaluation, and molecular dynamics simulations, we identified and characterized a novel small molecule, P10, which inhibits β-TrCP1/NRF2 interaction. The compound was validated using luciferase reporter assays, co-immunoprecipitation, and ubiquitination experiments. The specificity of P10 was assessed by comparing NRF2 signatures in wild-type and Nrf2-null cells. The impact of NRF2 activation induced by P10 was investigated by evaluating its antioxidant and anti-inflammatory responses against tert-butyl hydroperoxide and lipopolysaccharide, respectively. Finally, wild-type and Nrf2-null mice were administered P10 intraperitoneally at a dose of 20 mg/kg daily for five consecutive days. Four hours before sacrifice, all animals received a lipopolysaccharide (LPS) injection at 10 mg/kg.
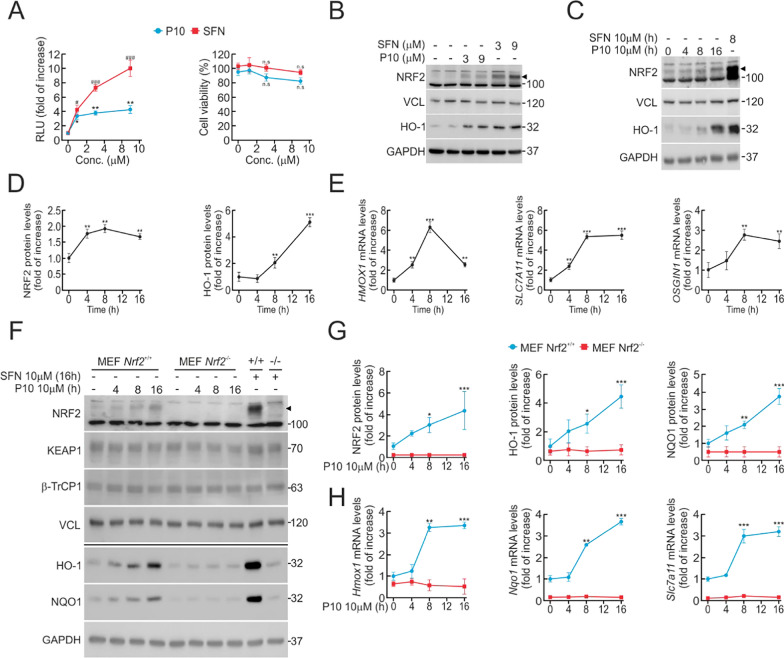
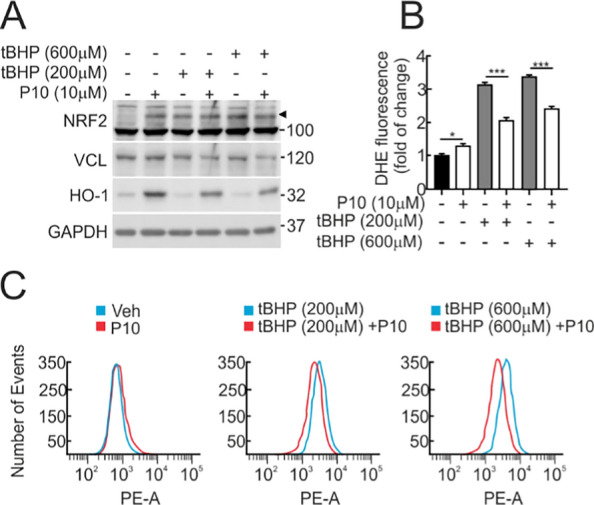
Results: P10 selectively disrupts the interaction between β-TrCP1 and NRF2, thereby inhibiting β-TrCP1-mediated ubiquitination of NRF2 and leading to the upregulation of NRF2 target genes. Additionally, P10 mitigates oxidative stress induced by tert-butyl hydroperoxide and reduces pro-inflammatory markers in an NRF2-dependent manner in macrophages treated with lipopolysaccharide. In a preclinical model of liver inflammation, P10 specifically targets the liver, significantly attenuating lipopolysaccharide-induced inflammation through the activation of NRF2. This is demonstrated by decreased expression of inflammatory cytokine genes and a reduction in F4/80-stained liver macrophages. Notably, this anti-inflammatory effect is absent in Nrf2-knockout mice, confirming its NRF2-dependent mechanism of action.
Conclusions: P10 emerges as a promising NRF2 activator by selectively disrupting the β-TrCP1/NRF2 interaction, highlighting its potential as a therapeutic agent for diseases presenting acute liver inflammation.
Keywords: Inflammation; Liver; NRF2; Protein–protein interaction-inhibitor; β-TrCP1.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing of interest. The funders had no role in the study's design; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures





References
-
- Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16(2):123–40. - PubMed
-
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. - PubMed
-
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
