

Transthyretin Amyloid Cardiomyopathy-2025 Update: Current Diagnostic Approaches and Emerging Therapeutic Options
- PMID: 40649158
- PMCID: PMC12250813
- DOI: 10.3390/jcm14134785
Transthyretin Amyloid Cardiomyopathy-2025 Update: Current Diagnostic Approaches and Emerging Therapeutic Options
Abstract
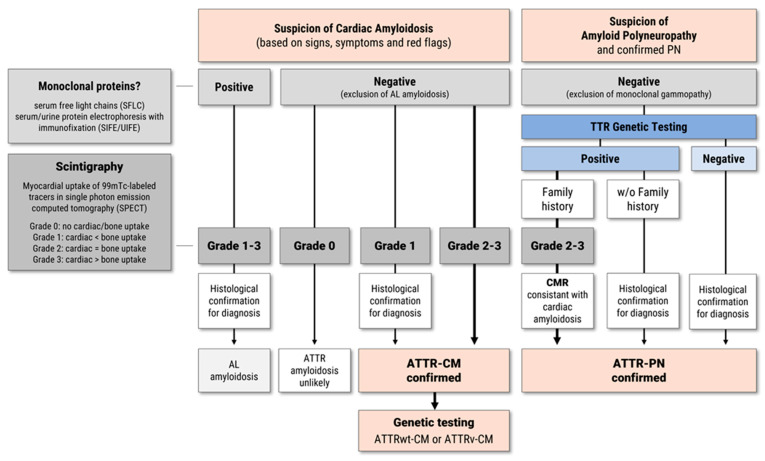
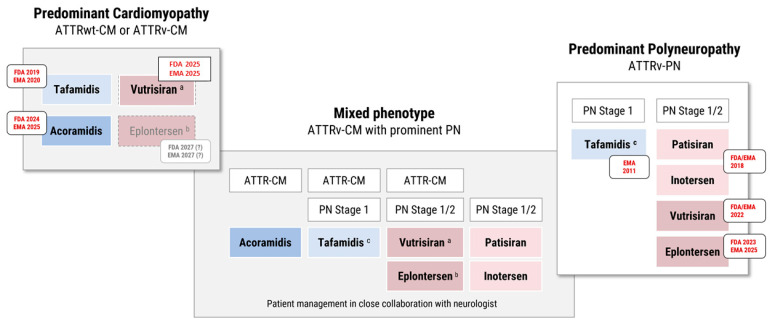
Transthyretin-related (ATTR) amyloidosis is a progressive, multisystem disease caused by the extracellular deposition of misfolded transthyretin (TTR) monomers as insoluble amyloid fibrils. Clinical manifestations vary widely and may include cardiomyopathy (ATTR-CM), polyneuropathy (ATTR-PN), or mixed phenotypes. The condition is increasingly recognized as an underdiagnosed contributor to heart failure, particularly in elderly patients. ATTR amyloidosis exists in two major forms: hereditary (ATTRv), resulting from mutations in the TTR gene, and wild-type (ATTRwt), typically affecting men over 70 years of age. Advances in disease understanding have led to a paradigm shift in management, with the introduction of targeted therapies that slow disease progression and improve prognosis. First-generation therapies such as tafamidis have demonstrated survival benefits in ATTR-CM. More recently, second-generation agents-such as the TTR stabilizer acoramidis and RNA silencers including vutrisiran and eplontersen-have shown promising efficacy in clinical trials. Additional strategies under investigation include gene editing and monoclonal antibodies targeting TTR amyloid deposits. This review outlines current diagnostic strategies and therapeutic options for ATTR amyloidosis, emphasizing the need for early detection and individualized treatment approaches. The expanding therapeutic landscape highlights the importance of accurate phenotyping and timely intervention to optimize clinical outcomes.
Keywords: ATTR cardiomyopathy; ATTR polyneuropathy; amyloidosis; antisense oligonucleotide; cardiac amyloidosis; clinical development; gene editing; siRNA; transthyretin.
Conflict of interest statement
C.T. has received speaker fees and/or contributions to congresses from Astra Zeneca, Bayer, Boehringer-Ingelheim, Novartis, and Pfizer. A.E. has no COI. A.V.K. has received honoraria from Alexion Pharmaceuticals, AstraZeneca, Attralus, Bayer Vital AG, and Bridgebio; honoraria and travel support from Akcea Therapeutics, Alnylam Pharmaceuticals, Alnylam Pharmaceuticals, and Pfizer Inc./Pharma GmbH; research support from Pfizer Inc./Pharma GmbH; was study investigator for Alnylam Pharmaceuticals (APOLLO, APOLLO OLE, ENDEAVOUR, HELIOS B) and IONIS Pharmaceuticals (NEURO-TTR); and advisory board member for Akcea Therapeutics, Alexion Pharmaceuticals, Alnylam Pharmaceuticals, AstraZeneca, Bayer Vital AG, Intellia Therapeutics, Novo Nordisk A/S, and Pfizer Inc./Pharma GmbH.
Figures

Similar articles
-
Advancing treatments for transthyretin amyloid cardiomyopathy: Innovations in RNA silencing, gene editing, TTR stabilization, and degradation.Kardiol Pol. 2025;83(2):121-137. doi: 10.33963/v.phj.104054. Epub 2025 Jan 7. Kardiol Pol. 2025. PMID: 39775625 Review.
-
Wild-Type Transthyretin Amyloid Depositions in the Subcutaneous Fat and Skeletal Muscles of a Nonagenarian Who Had Heart Failure With Preserved Ejection Fraction and No Myocardial Technetium-99m-Labeled Pyrophosphate Uptake.Cureus. 2025 May 24;17(5):e84759. doi: 10.7759/cureus.84759. eCollection 2025 May. Cureus. 2025. PMID: 40557028 Free PMC article.
-
Transthyretin Amyloid Cardiomyopathy: A Review of Approved Pharmacotherapies.Cardiol Rev. 2025 Jul 1. doi: 10.1097/CRD.0000000000000985. Online ahead of print. Cardiol Rev. 2025. PMID: 40590530
-
Transthyretin Kinetic Stabilizers for ATTR Amyloidosis: A Narrative Review of Mechanisms and Therapeutic Benefits.Cardiol Ther. 2025 Jul 29. doi: 10.1007/s40119-025-00423-7. Online ahead of print. Cardiol Ther. 2025. PMID: 40730935 Review.
-
Biomarkers in Subclinical Transthyretin Cardiac Amyloidosis.Curr Heart Fail Rep. 2025 Feb 13;22(1):8. doi: 10.1007/s11897-025-00696-y. Curr Heart Fail Rep. 2025. PMID: 39945945 Free PMC article. Review.
References
-
- Buxbaum J.N., Eisenberg D.S., Fandrich M., McPhail E.D., Merlini G., Saraiva M.J.M., Sekijima Y., Westermark P. Amyloid nomenclature 2024: Update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2024;31:249–256. doi: 10.1080/13506129.2024.2405948. - DOI - PubMed
-
- Gentile L., Coelho T., Dispenzieri A., Conceicao I., Waddington-Cruz M., Kristen A., Wixner J., Diemberger I., Gonzalez-Moreno J., Cariou E., et al. A 15-year consolidated overview of data in over 6000 patients from the Transthyretin Amyloidosis Outcomes Survey (THAOS) Orphanet J. Rare Dis. 2023;18:350. doi: 10.1186/s13023-023-02962-5. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
