

High mtDNA content identifies oxidative phosphorylation-driven acute myeloid leukemias and represents a therapeutic vulnerability
- PMID: 40653487
- PMCID: PMC12256626
- DOI: 10.1038/s41392-025-02303-x
High mtDNA content identifies oxidative phosphorylation-driven acute myeloid leukemias and represents a therapeutic vulnerability
Abstract
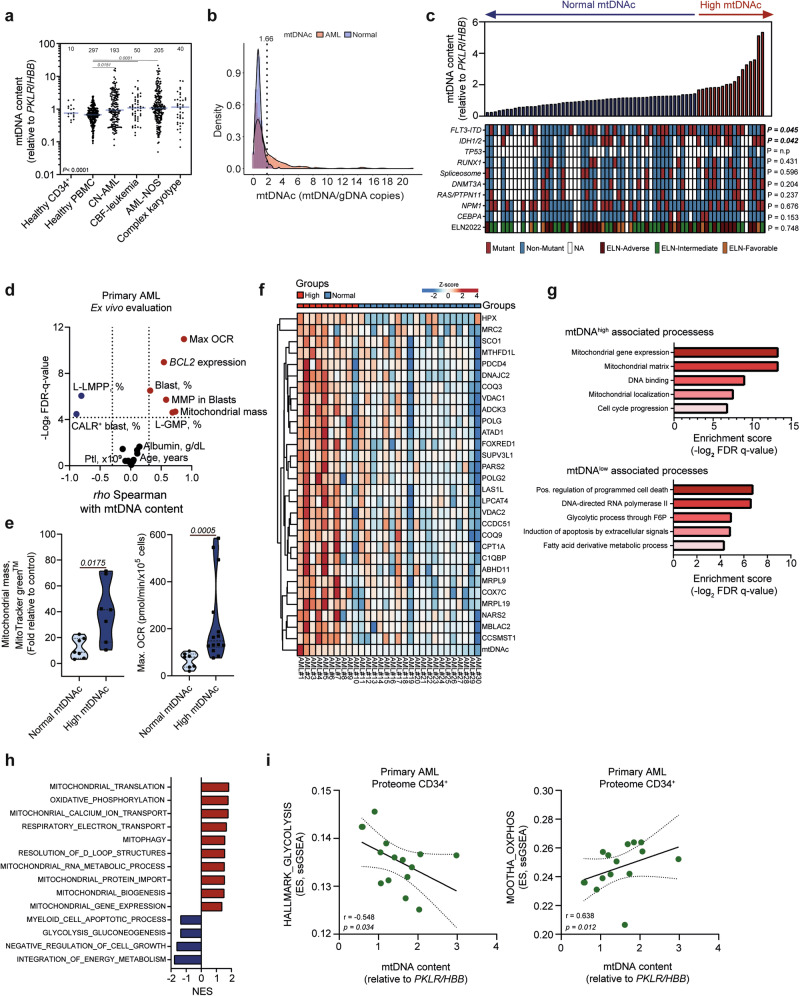
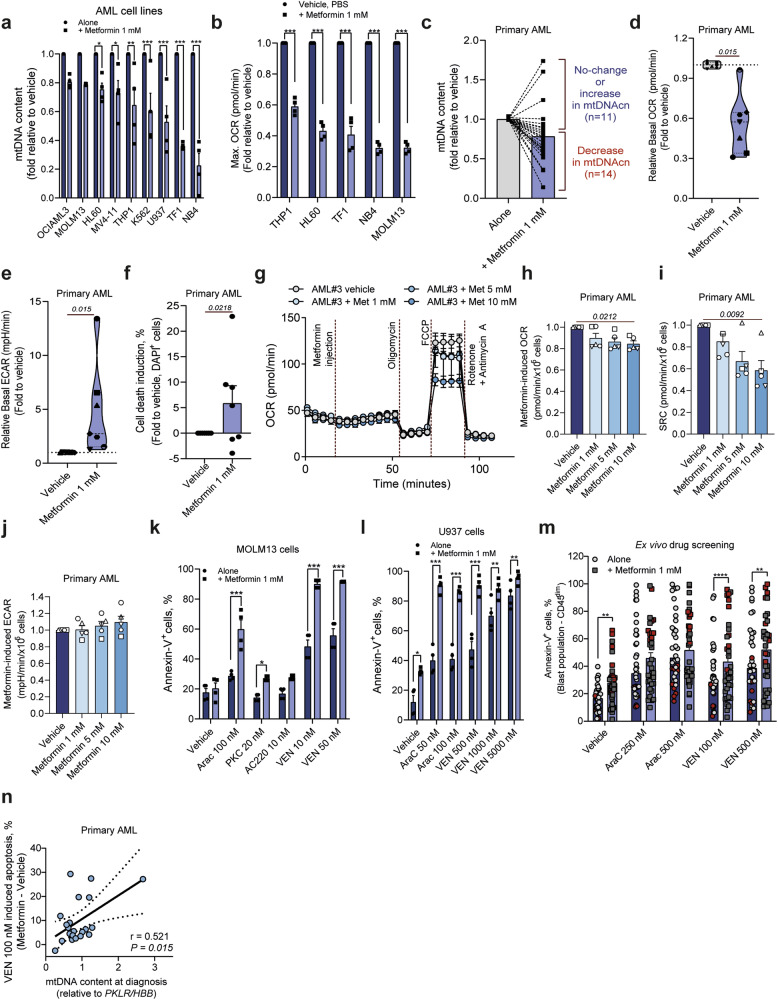
Metabolic reprogramming is a hallmark of cancer, with acute myeloid leukemia (AML) being no exception. Mitochondrial function, particularly its role in protecting tumor cells against chemotherapy, is of significant interest in AML chemoresistance. In this study, we identified mitochondrial DNA content (mtDNAc), measured by quantitative PCR, as a simple and precise marker to stratify the metabolic states of AML patients. We show that patients with high mtDNAc are associated with increased mitochondrial metabolism and a higher dependency on oxidative phosphorylation (OXPHOS), often correlating with chemoresistance. Clinically, patients receiving cytarabine and an anthracycline-based regimen (7 + 3 regimen) experienced inferior relapse-free survival and a higher overall rate of leukemia recurrence. Ex vivo experiments using primary AML samples confirmed cytarabine resistance in high mtDNAc patients, which could be overcome by inhibiting mitochondrial complex I. The FDA-approved drug metformin, which targets mitochondrial metabolism, significantly enhanced apoptosis in response to chemotherapy or targeted agents, such as venetoclax, in AML models. However, metformin-treated cells adapted by increasing glycolysis and NAD+ production, a resistance mechanism that could be bypassed by targeting the nicotinamide phosphoribosyltransferase (NAMPT) enzyme. In summary, we demonstrated that mtDNAc is an effective tool for assessing the metabolic state of AML cells. This method can be easily implemented in clinical practice to identify chemoresistant patients and guide personalized treatment strategies, including novel combination therapies for those with a high reliance on mitochondrial metabolism.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures


References
-
- Döhner, H., Weisdorf, D. J. & Bloomfield, C. D. Acute myeloid leukemia. N. Engl. J. Med.373, 1136–1152 (2015). - PubMed
-
- Baccelli, I. et al. Mubritinib targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia. Cancer Cell36, 84–99.e8 (2019). - PubMed
-
- Shallis, R. M. et al. Standardising acute myeloid leukaemia classification systems: a perspective from a panel of international experts. Lancet Haematol.10, e767–e776 (2023). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
