

Casein Kinase 2α Ablation Confers Protection Against Metabolic Dysfunction-Associated Steatotic Liver Disease: Role of FUN14 Domain Containing 1-Dependent Regulation of Mitophagy and Ferroptosis
- PMID: 40656543
- PMCID: PMC12246556
- DOI: 10.1002/mco2.70277
Casein Kinase 2α Ablation Confers Protection Against Metabolic Dysfunction-Associated Steatotic Liver Disease: Role of FUN14 Domain Containing 1-Dependent Regulation of Mitophagy and Ferroptosis
Abstract
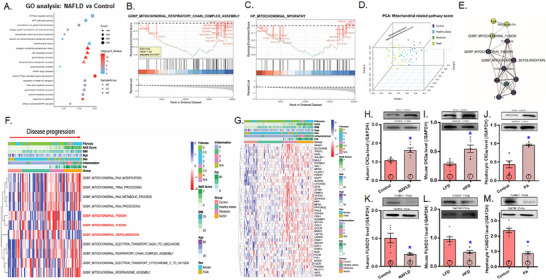
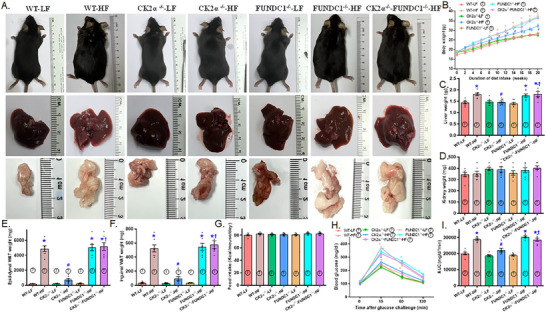
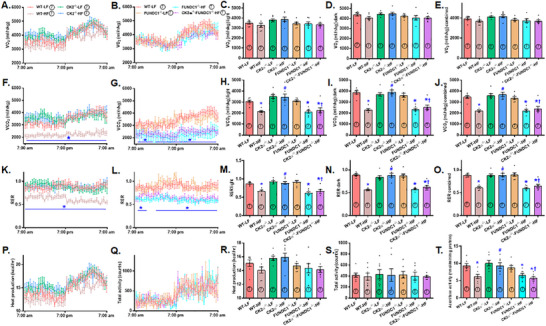
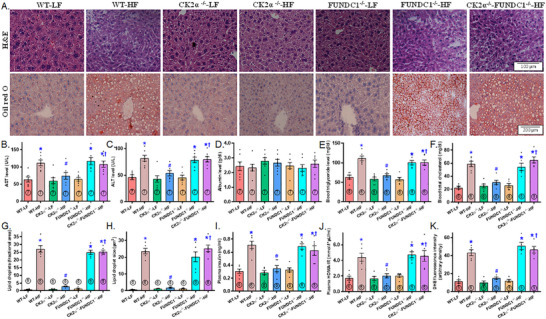
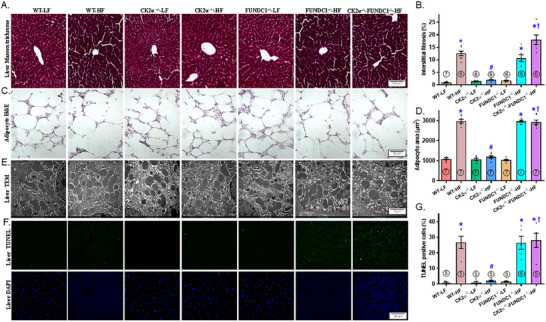
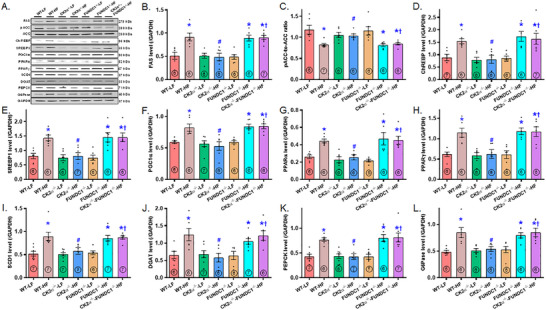
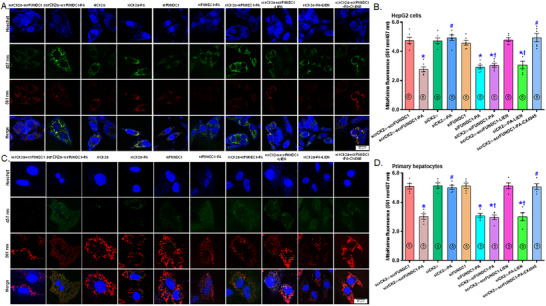
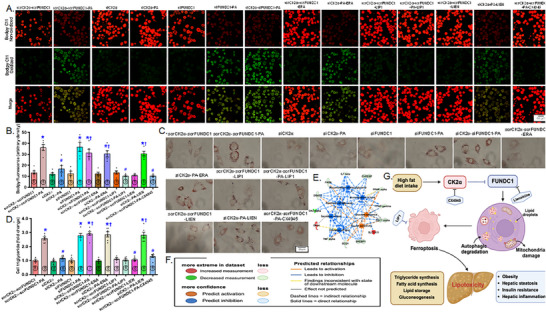
Mitochondrial dyshomeostasis provokes the onset of metabolic dysfunction-associated steatotic liver disease (MASLD) although its precise involvement in particular mitophagy in MASLD remains elusive. This work evaluated the role of casein kinase 2α (CK2α) and FUNDC1 in high-fat diet (HFD)-evoked MASLD. WT and CK2α deletion (CK2α -/- ) mice were subjected to low fat or HFD for 20 weeks. Global metabolism, AST, ALT, cholesterol, triglycerides, hepatic steatosis, fibrosis, inflammation, mitochondrial injury, mitophagy and ferroptosis were examined. Bioinformatics analysis enriched mitochondria-related pathways in MASLD. Hepatic CK2α and FUNDC1 were upregulated and downregulated, respectively, in MASLD patients and HFD-fed mice. HFD led to adiposity, hepatomegaly, hepatic steatosis, fibrosis, inflammation, ferroptosis, mitochondrial injury, elevated hepatic tissue Fe2+, FAS, CHREBP, SREBP1, PGC1α, PPARα, PPARγ, SCD1, PEPCK, G6Pase, and DGAT1 as well as downregulated FUNDC1, GPx4, SLC7A11 and NCOA4, the effects (except for NCOA4) were nullified by CK2α deletion. FUNDC1 deletion nullified CK2α deletion-evoked benefit on hepatic ferroptosis and lipid enzymes. In vitro study using palmitic acid indicated an obligatory role for CK2α, FUNDC1 and ferroptosis in hepatocyte steatosis. Collectively, our results demonstrated that CK2α activation by HFD serves as a trigger for mitochondrial damage, hepatic injury, and pathogenesis of MASLD through FUNDC1 disruption and ferroptosis.
Keywords: FUN14 domain containing 1; MASLD; casein kinase 2α; ferroptosis; steatosis.
© 2025 The Author(s). MedComm published by Sichuan International Medical Exchange & Promotion Association (SCIMEA) and John Wiley & Sons Australia, Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Ghrelin attenuates metabolic dysfunction-associated steatotic liver disease by activating glutamine metabolism via inhibition of the IRE1α-XBP-1 pathway.Biochem Pharmacol. 2025 Oct;240:117089. doi: 10.1016/j.bcp.2025.117089. Epub 2025 Jun 26. Biochem Pharmacol. 2025. PMID: 40581331
-
Hepatocyte-specific RIG-I loss attenuates metabolic dysfunction-associated steatotic liver disease in mice via changes in mitochondrial respiration and metabolite profiles.Toxicol Res. 2024 Sep 23;40(4):683-695. doi: 10.1007/s43188-024-00264-x. eCollection 2024 Oct. Toxicol Res. 2024. PMID: 39345739
-
Activation of TRPA1 prevents metabolic dysfunction-associated steatotic liver disease in diet-induced obese mice through stimulating the AMPK/CPT1A signaling pathway.J Physiol Biochem. 2025 May;81(2):359-373. doi: 10.1007/s13105-025-01081-y. Epub 2025 Apr 24. J Physiol Biochem. 2025. PMID: 40272632
-
Inappropriate Diet Exacerbates Metabolic Dysfunction-Associated Steatotic Liver Disease via Abdominal Obesity.Nutrients. 2024 Dec 5;16(23):4208. doi: 10.3390/nu16234208. Nutrients. 2024. PMID: 39683601 Free PMC article.
-
Food Nutrients and Bioactive Compounds for Managing Metabolic Dysfunction-Associated Steatotic Liver Disease: A Comprehensive Review.Nutrients. 2025 Jul 3;17(13):2211. doi: 10.3390/nu17132211. Nutrients. 2025. PMID: 40647314 Free PMC article. Review.
References
-
- Stefan N., Yki‐Jarvinen H., and Neuschwander‐Tetri B. A., “Metabolic Dysfunction‐Associated Steatotic Liver Disease: Heterogeneous Pathomechanisms and Effectiveness of Metabolism‐Based Treatment,” Lancet Diabetes & Endocrinology 13, no. 2 (2024): P134–148. - PubMed
-
- Do A., Zahrawi F., and Mehal W. Z., “Therapeutic Landscape of Metabolic Dysfunction‐Associated Steatohepatitis (MASH),” Nature Reviews Drug Discovery 24, no. 3 (2024): 171–189. - PubMed
-
- Wong R. J., Liu B., and Bhuket T., “Significant Burden of Nonalcoholic Fatty Liver Disease With Advanced Fibrosis in the US: A Cross‐Sectional Analysis of 2011–2014 National Health and Nutrition Examination Survey,” Alimentary Pharmacology & Therapeutics 46, no. 10 (2017): 974–980. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous