

Harnessing AlphaFold to reveal hERG channel conformational state secrets
- PMID: 40658102
- PMCID: PMC12259024
- DOI: 10.7554/eLife.104901
Harnessing AlphaFold to reveal hERG channel conformational state secrets
Abstract
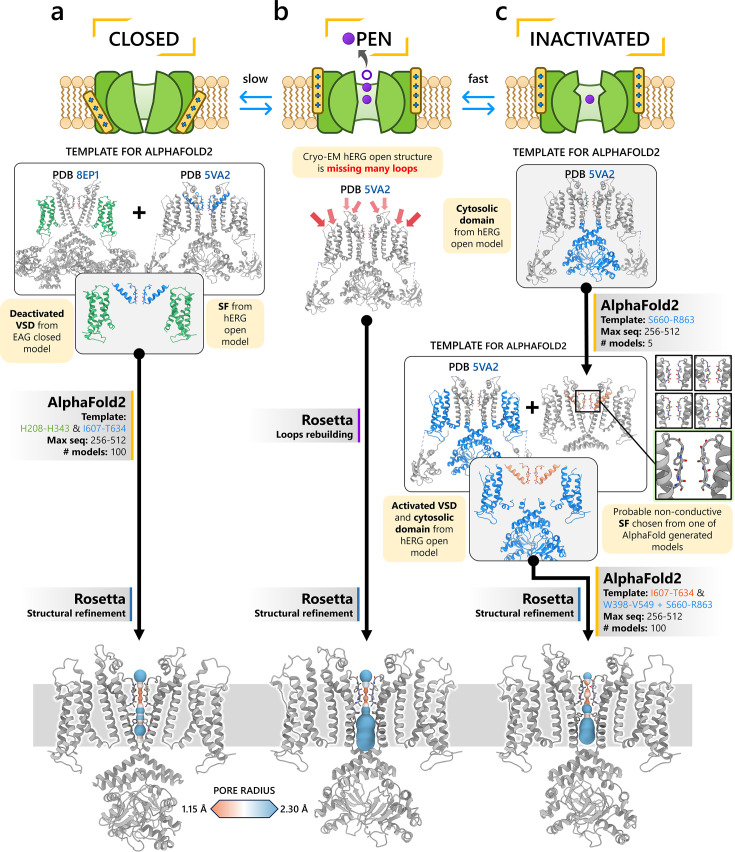
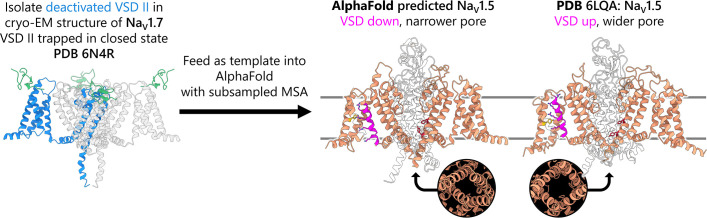
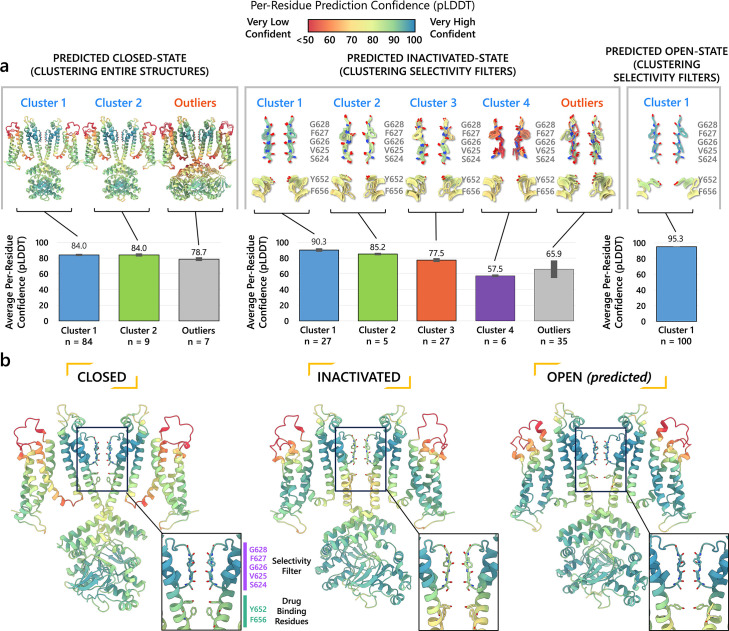
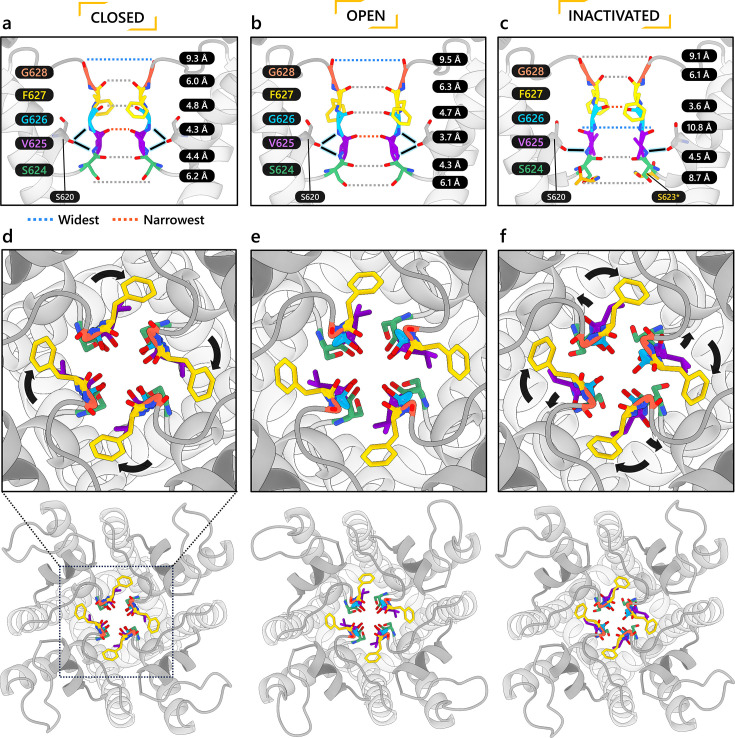
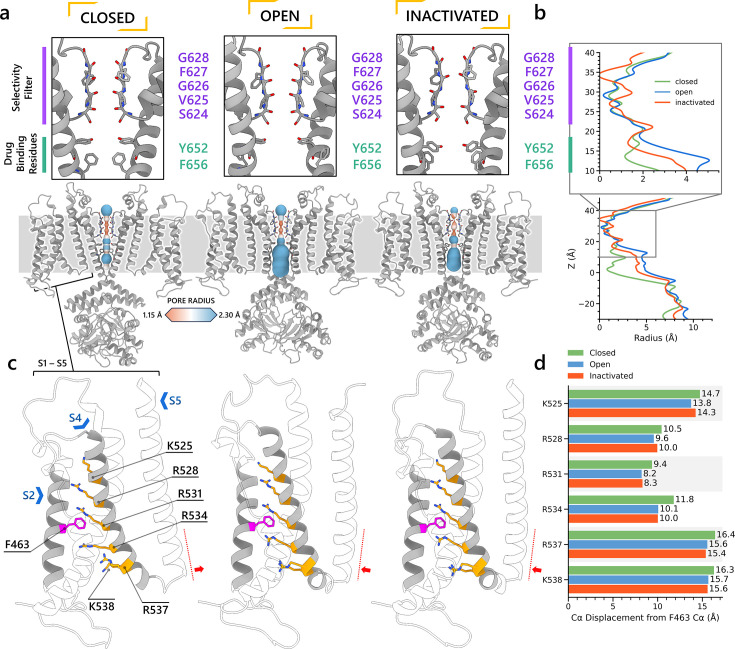
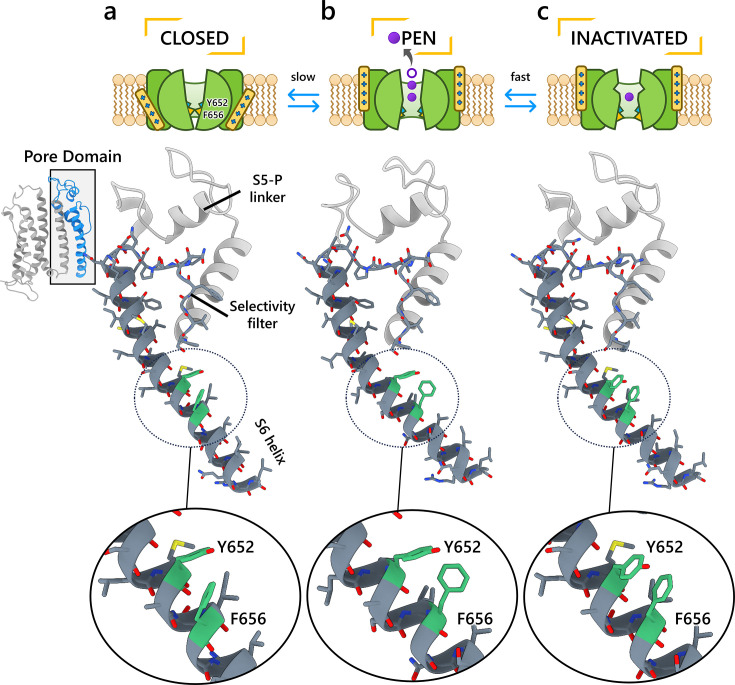
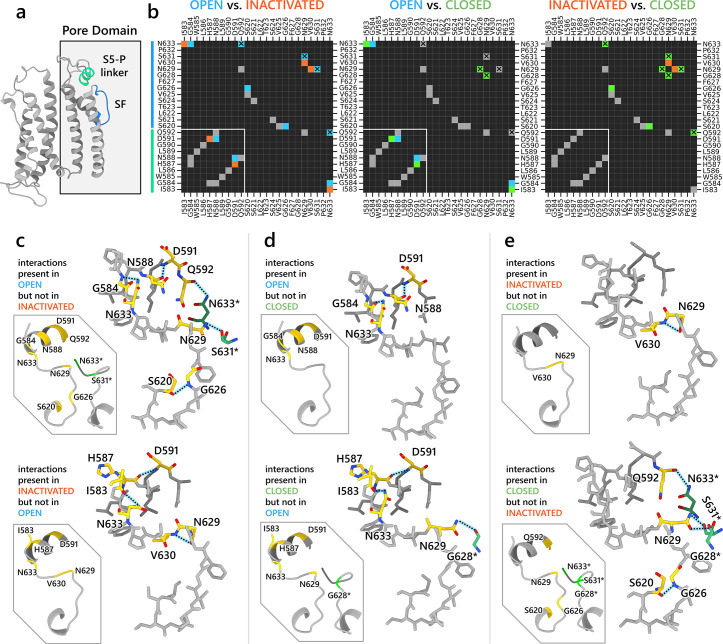
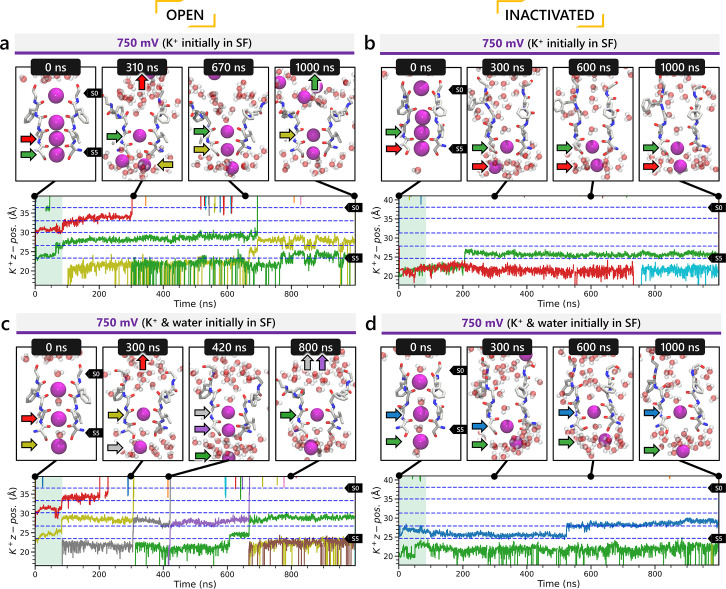
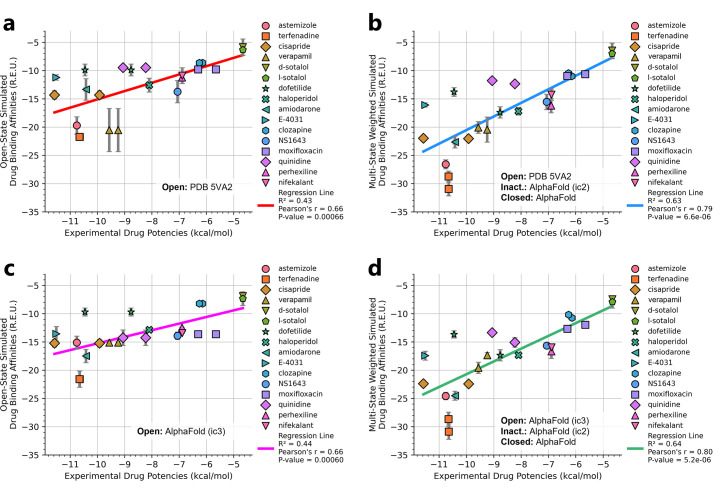
To design safe, selective, and effective new therapies, there must be a deep understanding of the structure and function of the drug target. One of the most difficult problems to solve has been the resolution of discrete conformational states of transmembrane ion channel proteins. An example is KV11.1 (hERG), comprising the primary cardiac repolarizing current, Ikr. hERG is a notorious drug anti-target against which all promising drugs are screened to determine potential for arrhythmia. Drug interactions with the hERG inactivated state are linked to elevated arrhythmia risk, and drugs may become trapped during channel closure. While prior studies have applied AlphaFold to predict alternative protein conformations, we show that the inclusion of carefully chosen structural templates can guide these predictions toward distinct functional states. This targeted modeling approach is validated through comparisons with experimental data, including proposed state-dependent structural features, drug interactions from molecular docking, and ion conduction properties from molecular dynamics simulations. Remarkably, AlphaFold not only predicts inactivation mechanisms of the hERG channel that prevent ion conduction but also uncovers novel molecular features explaining enhanced drug binding observed during inactivation, offering a deeper understanding of hERG channel function and pharmacology. Furthermore, leveraging AlphaFold-derived states enhances computational screening by significantly improving agreement with experimental drug affinities, an important advance for hERG as a key drug safety target where traditional single-state models miss critical state-dependent effects. By mapping protein residue interaction networks across closed, open, and inactivated states, we identified critical residues driving state transitions validated by prior mutagenesis studies. This innovative methodology sets a new benchmark for integrating deep learning-based protein structure prediction with experimental validation. It also offers a broadly applicable approach using AlphaFold to predict discrete protein conformations, reconcile disparate data, and uncover novel structure-function relationships, ultimately advancing drug safety screening and enabling the design of safer therapeutics.
Keywords: AlphaFold; arrhythmia; biochemistry; chemical biology; hERG; human; molecular biophysics; molecular docking; molecular dynamics simulation; structural biology; voltage-gated potassium channel.
© 2024, Ngo et al.
Conflict of interest statement
KN, PY, VY, CC, IV No competing interests declared
Figures


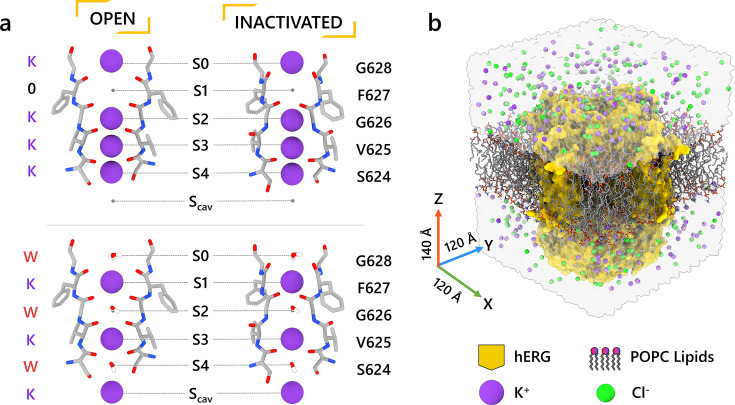
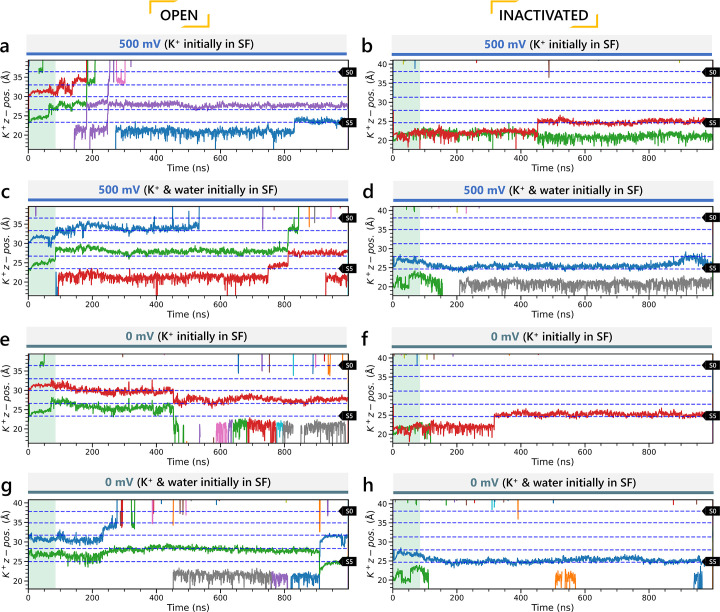

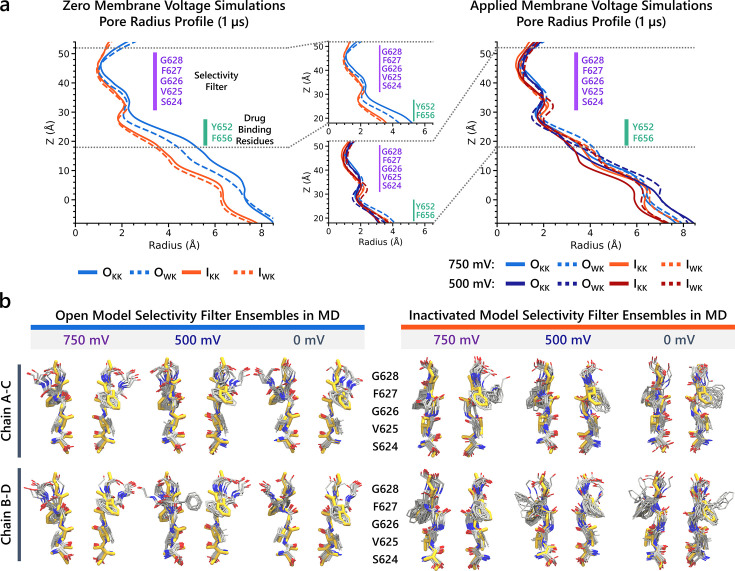
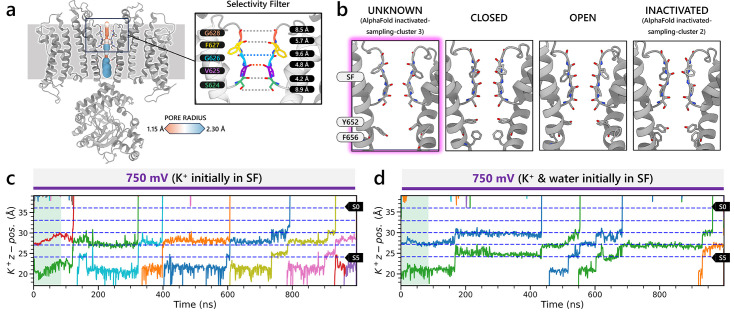
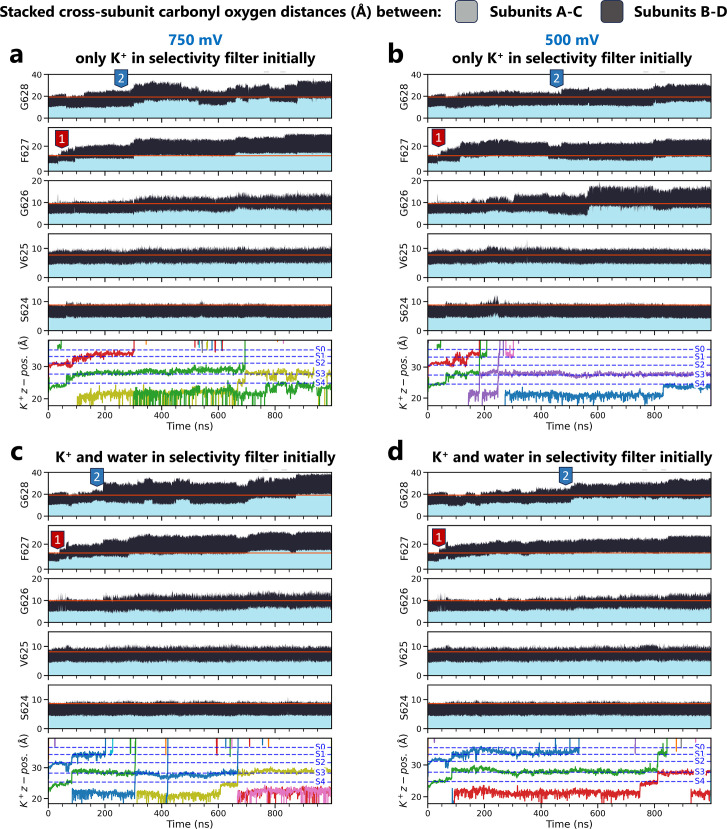
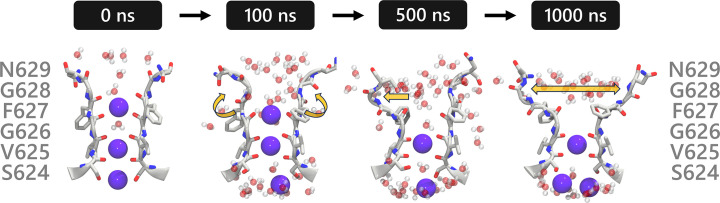
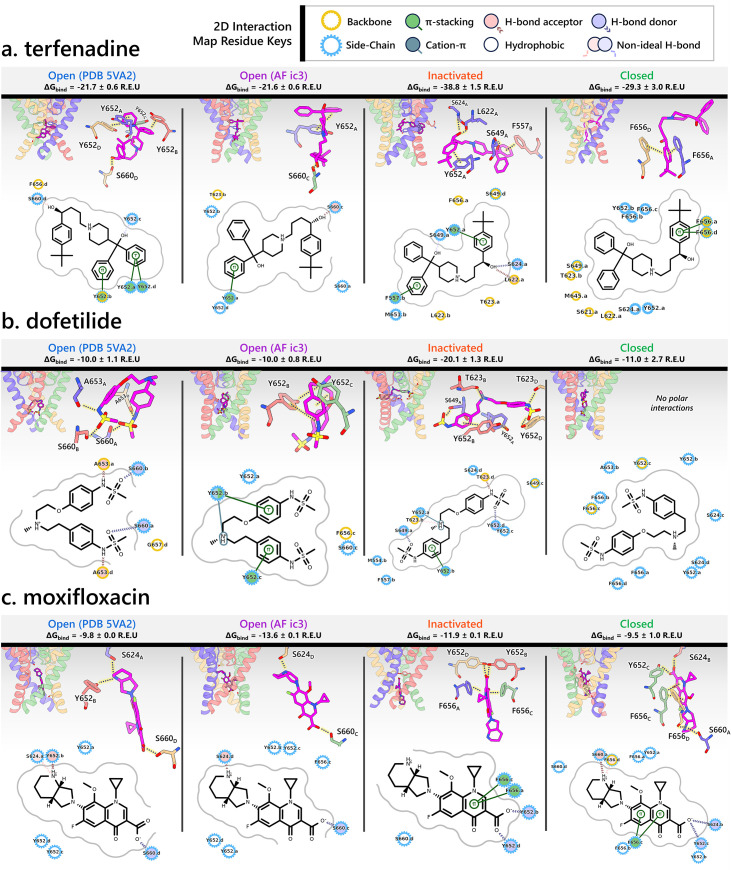
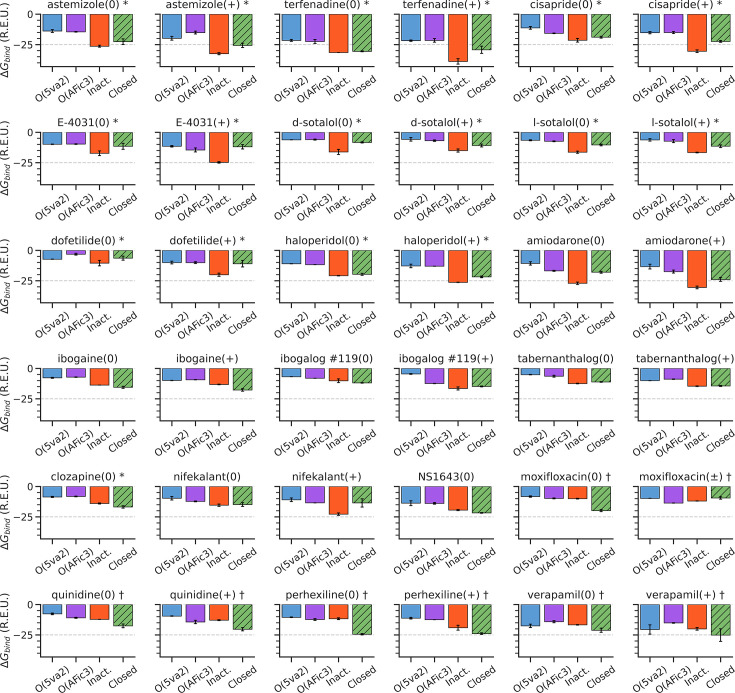

Update of
-
Harnessing AlphaFold to reveal hERG channel conformational state secrets.bioRxiv [Preprint]. 2025 Apr 15:2024.01.27.577468. doi: 10.1101/2024.01.27.577468. bioRxiv. 2025. Update in: Elife. 2025 Jul 14;13:RP104901. doi: 10.7554/eLife.104901. PMID: 38352360 Free PMC article. Updated. Preprint.
Similar articles
-
Harnessing AlphaFold to reveal hERG channel conformational state secrets.bioRxiv [Preprint]. 2025 Apr 15:2024.01.27.577468. doi: 10.1101/2024.01.27.577468. bioRxiv. 2025. Update in: Elife. 2025 Jul 14;13:RP104901. doi: 10.7554/eLife.104901. PMID: 38352360 Free PMC article. Updated. Preprint.
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
Cited by
-
T-World: A highly general computational model of a human ventricular myocyte.bioRxiv [Preprint]. 2025 Mar 28:2025.03.24.645031. doi: 10.1101/2025.03.24.645031. bioRxiv. 2025. PMID: 40196542 Free PMC article. Preprint.
References
-
- Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, Bodenstein SW, Evans DA, Hung C-C, O’Neill M, Reiman D, Tunyasuvunakool K, Wu Z, Žemgulytė A, Arvaniti E, Beattie C, Bertolli O, Bridgland A, Cherepanov A, Congreve M, Cowen-Rivers AI, Cowie A, Figurnov M, Fuchs FB, Gladman H, Jain R, Khan YA, Low CMR, Perlin K, Potapenko A, Savy P, Singh S, Stecula A, Thillaisundaram A, Tong C, Yakneen S, Zhong ED, Zielinski M, Žídek A, Bapst V, Kohli P, Jaderberg M, Hassabis D, Jumper JM. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493–500. doi: 10.1038/s41586-024-07487-w. - DOI - PMC - PubMed
-
- Asai T, Adachi N, Moriya T, Oki H, Maru T, Kawasaki M, Suzuki K, Chen S, Ishii R, Yonemori K, Igaki S, Yasuda S, Ogasawara S, Senda T, Murata T. Cryo-EM Structure of K+-Bound hERG channel complexed with the Blocker Astemizole. Structure. 2021;29:203–212. doi: 10.1016/j.str.2020.12.007. - DOI - PubMed
MeSH terms
Substances
Associated data
Grants and funding
- MCB20010/Texas Advanced Computing Center (TACC)
- U01HL126273/HL/NHLBI NIH HHS/United States
- R01 HL128537/HL/NHLBI NIH HHS/United States
- T32 HL086350/HL/NHLBI NIH HHS/United States
- 2032486/National Science Foundation
- R01HL085844/HL/NHLBI NIH HHS/United States
- PSCA17085P/Pittsburgh Supercomputing Center (PSC)
- PSCA16108P/Pittsburgh Supercomputing Center (PSC)
- R01 HL174001/HL/NHLBI NIH HHS/United States
- R01 HL085844/HL/NHLBI NIH HHS/United States
- Department of Physiology and Membrane Biology Research Partnership Fund/University of California Davis School of Medicine
- MCB170095/Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS)
- OT2OD026580/NH/NIH HHS/United States
- PSCA18077P/Pittsburgh Supercomputing Center (PSC)
- MCB160089P/Pittsburgh Supercomputing Center (PSC)
- OT2 OD026580/OD/NIH HHS/United States
- R01HL128537/HL/NHLBI NIH HHS/United States
- R01 HL152681/HL/NHLBI NIH HHS/United States
- Oracle for Research fellowship/Oracle
- T32HL086350/HL/NHLBI NIH HHS/United States
- 19CDA34770101/American Heart Association
- U01 HL126273/HL/NHLBI NIH HHS/United States
- R01 GM116961/GM/NIGMS NIH HHS/United States
- R01HL152681/HL/NHLBI NIH HHS/United States
- R01HL174001/HL/NHLBI NIH HHS/United States
