

T cell-specific non-viral DNA delivery and in vivo CAR-T generation using targeted lipid nanoparticles
- PMID: 40659448
- PMCID: PMC12258353
- DOI: 10.1136/jitc-2025-011759
T cell-specific non-viral DNA delivery and in vivo CAR-T generation using targeted lipid nanoparticles
Abstract
Background: Ex vivo chimeric antigen receptor (CAR)-T therapies have revolutionized cancer treatment. However, treatment accessibility is hindered by high costs, long manufacturing times, and the need for specialized centers and inpatient care. Strategies to generate CAR-T cells in vivo have emerged as a promising alternative that could bypass CAR-T manufacturing bottlenecks. Most current in vivo CAR-T approaches, while demonstrating encouraging preclinical efficacy, rely on transient messenger RNA (mRNA) delivery or viral vectors which both have limitations in terms of efficiency, durability, and scalability. To address these challenges, we developed a novel DNA-based targeted lipid nanoparticle (LNP) which we termed NCtx.
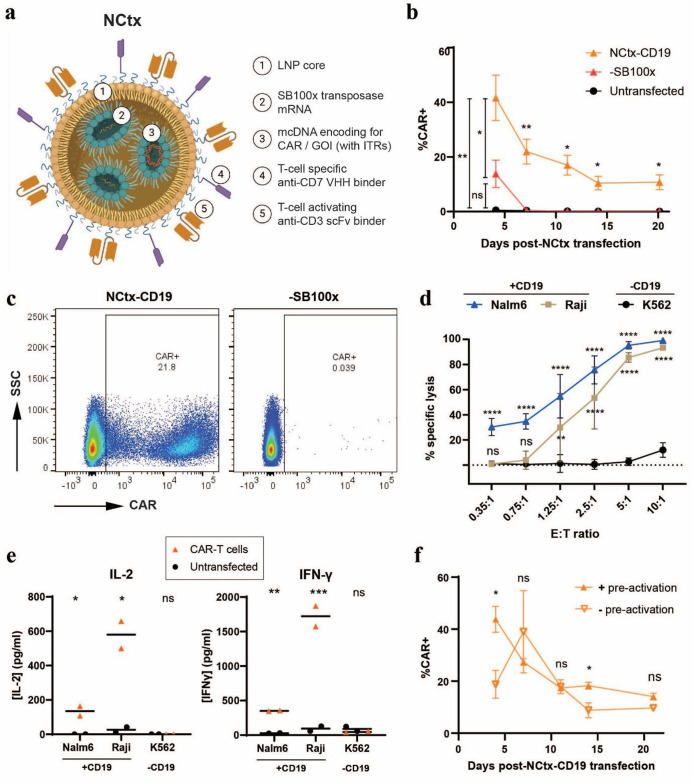
Methods: Minicircle DNA (mcDNA) encoding a CAR construct and SB100x transposase mRNA were encapsulated within a novel lipid formulation which was functionalized with T cell-specific anti-CD7 and anti-CD3 binders. In vitro, we evaluated T cell specificity, mcDNA and mRNA transfection efficiency, transposon-mediated CAR integration and functionality of the resulting CAR-T cells. In vivo efficacy was assessed in peripheral blood mononuclear cell and CD34+ stem cell humanized murine xenograft models of B cell leukemia.
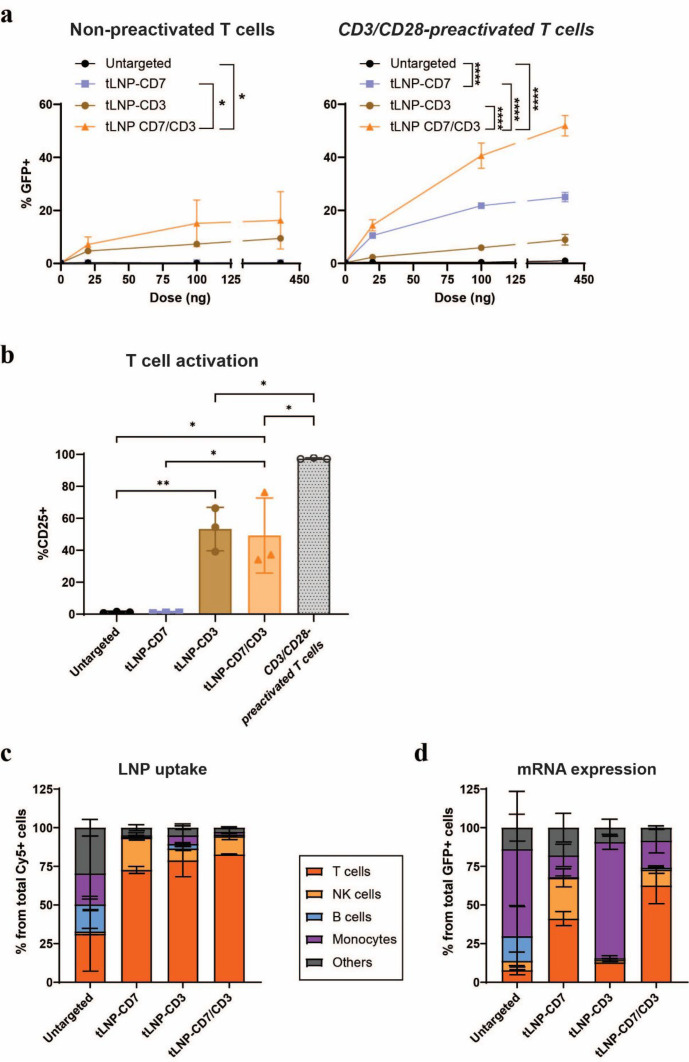
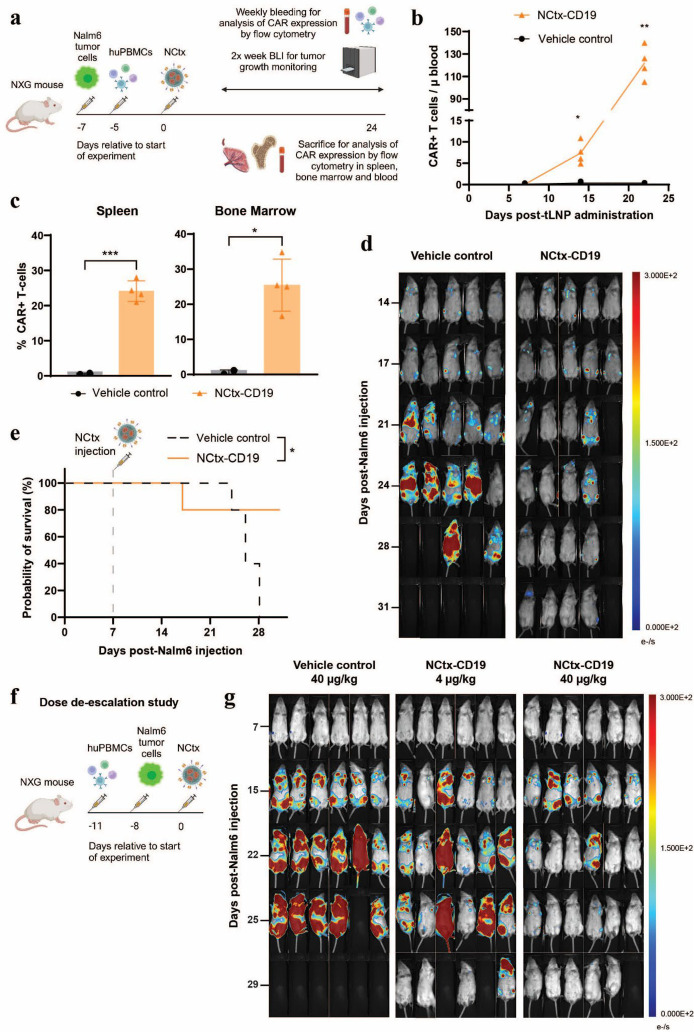
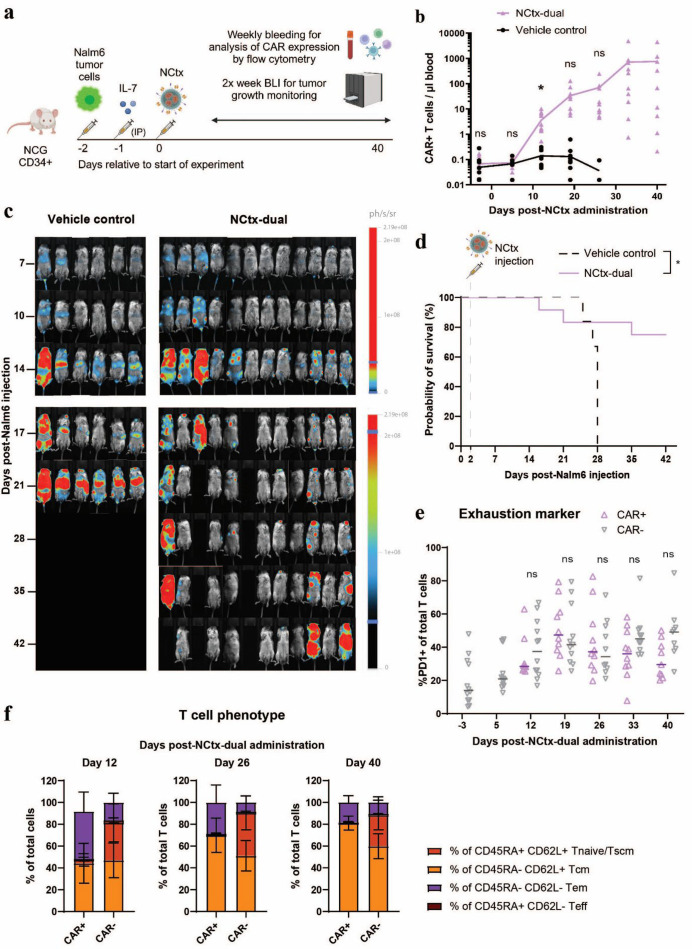
Results: In vitro, NCtx displayed high specificity and transfection efficiency with both mcDNA and mRNA in primary T cells. Transposase mRNA facilitated genomic integration of the CAR gene, leading to the generation of stable CAR-T cells that exhibited antigen-specific cytotoxicity and cytokine release. In vivo, a single intravenous dose of NCtx induced robust CAR-T cell generation resulting in effective tumor control and significantly improved survival in two distinct xenograft models.
Conclusions: Our findings demonstrate for the first time that targeted LNPs can be employed for efficient DNA delivery to T cells in vitro and in vivo. We show that when combined with transposase technology, this LNP-based system can efficiently generate stable CAR-T cells directly in vivo, inducing potent and durable antitumor responses. NCtx represents a novel non-viral gene therapy vector for in vivo CAR-T therapy, offering a scalable and potentially more accessible alternative to traditional approaches in CAR-T cell generation.
Keywords: Chimeric antigen receptor - CAR; Gene therapy; Immunotherapy; Nanoparticle; T cell.
© Author(s) (or their employer(s)) 2025. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ Group.
Conflict of interest statement
Competing interests: All authors are either full-time or part-time employees of NanoCell Therapeutics and hold equity or stock options in the company. MG, JL, EvD, DM, AA, ME, SN, NS and ZL are authors on patent applications related to this work. PM and MG are founders of Supercoiled GeneTx GmbH, which owns intellectual property rights to the minicircle DNA technology used in this study.
Figures
References
-
- Ahmed N, Sun F, Teigland C, et al. Chimeric Antigen Receptor T-Cell Access in Patients with Relapsed/Refractory Large B-Cell Lymphoma: Association of Access with Social Determinants of Health and Travel Time to Treatment Centers. Transplantation and Cellular Therapy . 2024;30:714–25. doi: 10.1016/j.jtct.2024.04.017. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources