

Uncovering methylation-dependent genetic effects on regulatory element function in diverse genomes
- PMID: 40659498
- PMCID: PMC12315712
- DOI: 10.1101/gr.279957.124
Uncovering methylation-dependent genetic effects on regulatory element function in diverse genomes
Abstract
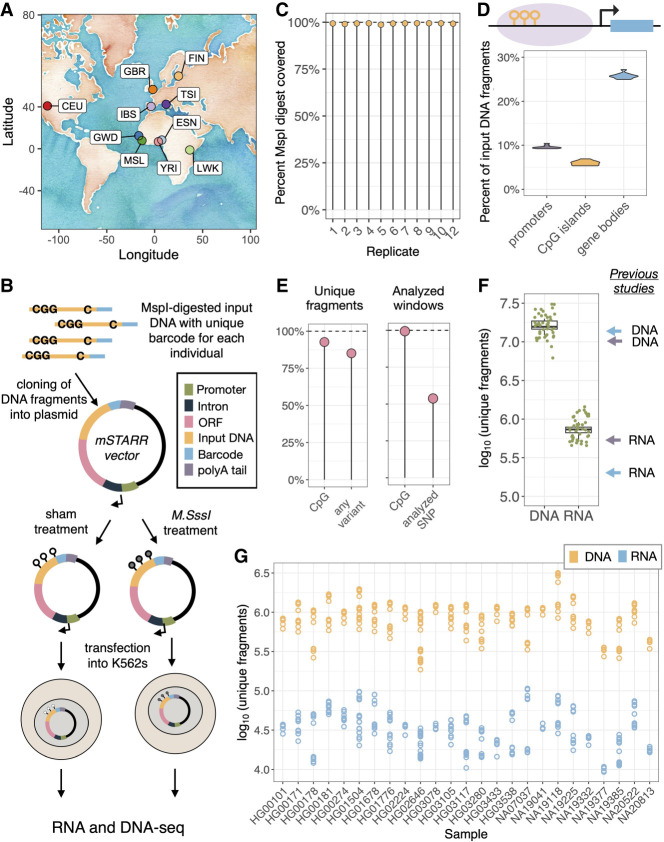
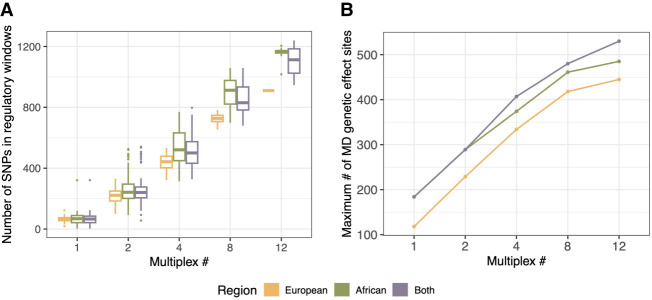
A major goal in evolutionary biology and biomedicine is to understand the complex interactions between genetic variants, the epigenome, and gene expression. However, the causal relationships between these factors remain poorly understood. mSTARR-seq, a methylation-sensitive massively parallel reporter assay, is capable of identifying methylation-dependent regulatory activity at many thousands of genomic regions simultaneously and allows for the testing of causal relationships between DNA methylation and gene expression on a region-by-region basis. Here, we develop a multiplexed mSTARR-seq protocol to assay naturally occurring human genetic variation from 25 individuals from 10 localities in Europe and Africa. We identify 6957 regulatory elements in either the unmethylated or methylated state, and this set was enriched for enhancer and promoter chromatin annotations, as expected. The expression of 58% of these regulatory elements is modulated by methylation, which is generally associated with decreased transcription. Within our set of regulatory elements, we use allele-specific expression analyses to identify 8020 sites with genetic effects on gene regulation; further, we find that 42.3% of these genetic effects vary in direction or magnitude between methylated and unmethylated states. Sites exhibiting methylation-dependent genetic effects are enriched for GWAS and EWAS annotations, implicating them in human disease. Compared with data sets that assay DNA from a single European ancestry individual, our multiplexed assay is able to uncover more genetic effects and methylation-dependent genetic effects, highlighting the importance of including diverse genomes in assays that aim to understand gene regulatory processes.
© 2025 Petersen et al.; Published by Cold Spring Harbor Laboratory Press.
Figures



Update of
-
Uncovering methylation-dependent genetic effects on regulatory element function in diverse genomes.bioRxiv [Preprint]. 2024 Aug 24:2024.08.23.609412. doi: 10.1101/2024.08.23.609412. bioRxiv. 2024. Update in: Genome Res. 2025 Aug 1;35(8):1781-1793. doi: 10.1101/gr.279957.124. PMID: 39229133 Free PMC article. Updated. Preprint.
Similar articles
-
Uncovering methylation-dependent genetic effects on regulatory element function in diverse genomes.bioRxiv [Preprint]. 2024 Aug 24:2024.08.23.609412. doi: 10.1101/2024.08.23.609412. bioRxiv. 2024. Update in: Genome Res. 2025 Aug 1;35(8):1781-1793. doi: 10.1101/gr.279957.124. PMID: 39229133 Free PMC article. Updated. Preprint.
-
The effect of sample site and collection procedure on identification of SARS-CoV-2 infection.Cochrane Database Syst Rev. 2024 Dec 16;12(12):CD014780. doi: 10.1002/14651858.CD014780. Cochrane Database Syst Rev. 2024. PMID: 39679851 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2020 Jan 9;1(1):CD011535. doi: 10.1002/14651858.CD011535.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2021 Apr 19;4:CD011535. doi: 10.1002/14651858.CD011535.pub4. PMID: 31917873 Free PMC article. Updated.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
High-throughput library transgenesis in Caenorhabditis elegans via Transgenic Arrays Resulting in Diversity of Integrated Sequences (TARDIS).Elife. 2023 Jul 4;12:RP84831. doi: 10.7554/eLife.84831. Elife. 2023. PMID: 37401921 Free PMC article.
References
-
- Banovich NE, Lan X, McVicker G, Van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. 2014. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet 10: e1004663. 10.1371/journal.pgen.1004663 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources