

Epigenetic regulation of MED12: a key contributor to the leukemic chromatin landscape and transcriptional dysregulation
- PMID: 40660382
- PMCID: PMC12261745
- DOI: 10.1186/s13072-025-00610-9
Epigenetic regulation of MED12: a key contributor to the leukemic chromatin landscape and transcriptional dysregulation
Abstract
Background: MED12 is a key regulator of transcription and chromatin architecture, essential for normal hematopoiesis. While its dysregulation has been implicated in hematological malignancies, the mechanisms driving its upregulation in acute myeloid leukemia (AML) remain poorly understood. We investigated MED12 expression across AML subgroups by integrating chromatin accessibility profiling, histone modification landscapes, and DNA methylation (DNAm) patterns. Functional assays using DNMT inhibition were performed to dissect the underlying regulatory mechanisms.
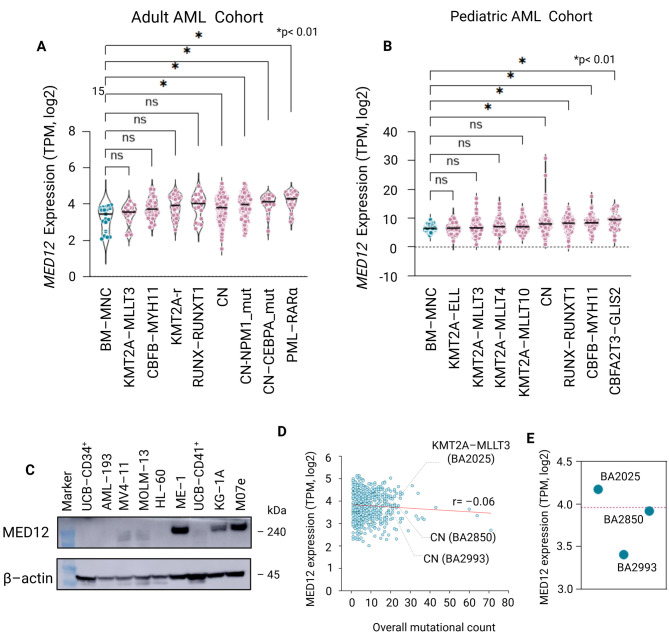
Results: MED12 shows subtype-specific upregulation in AML compared to hematopoietic stem and progenitor cells, independent of somatic mutations. Chromatin accessibility profiling reveals that the MED12 locus is epigenetically primed in AML blasts, with increased DNase hypersensitivity at regulatory elements. Histone modification analysis demonstrates strong H3K4me3 and H3K27ac enrichment around the transcription start site (TSS), consistent with promoter activation, while upstream and intragenic regions exhibit enhancer-associated marks (H3K4me1, H3K27ac). Notably, hypermethylation within TSS-proximal regulatory regions (TPRRs)-including promoter-overlapping and adjacent CpG islands-correlates with ectopic MED12 overexpression, challenging the canonical view of DNAm as strictly repressive. Functional studies show that DNMT inhibition via 5-azacytidine reduces MED12 expression despite promoter demethylation in cells with hypermethylated TPRRs, suggesting a noncanonical role for DNA methylation in maintaining active transcription. Furthermore, MED12 expression positively correlates with DNMT3A and DNMT3B expression, implicating these methyltransferases in sustaining its epigenetic activation.
Conclusion: This study identifies a novel regulatory axis in which aberrant DNA methylation, rather than genetic mutation, drives MED12 upregulation in AML. Our findings suggest that TPRR hypermethylation may function noncanonically to support transcriptional activation, likely in cooperation with enhancer elements. These results underscore the importance of epigenetic mechanisms in AML and highlight enhancer-linked methylation as a potential contributor to oncogene dysregulation. Future studies should further explore the role of noncanonical methylation-mediated gene activation in AML pathogenesis and therapeutic targeting.
Keywords: Acute myeloid leukemia; DNA methylation; Enhancer; MED12.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Transcriptome (mRNA expression) data analysis was conducted on pAML specimens from consented patients enrolled in the TARGET-AML study (phs000465), with access granted via the Database of Genotypes and Phenotypes (dbGaP). Transcriptome and mutation data for adult AML samples were obtained from the BEAT AML 2.0 study through VIZOME. Data on DNase hypersensitivity, histone modifications, and DNA methylation for primary AML patients were accessed from the BLUEPRINT Epigenome Consortium. Consent for publication: All authors have read the final version of manuscript and consent for publication. Competing interests: The authors declare no competing interests.
Figures




Similar articles
-
The loss of hepatitis B virus receptor NTCP/SLC10A1 in human liver cancer cells is due to epigenetic silencing.J Virol. 2024 Oct 22;98(10):e0118724. doi: 10.1128/jvi.01187-24. Epub 2024 Sep 19. J Virol. 2024. PMID: 39297647 Free PMC article.
-
The transcription factor HOXA9 induces expression of the chromatin modifier SMYD3 to drive leukemogenesis.J Biol Chem. 2025 Jul;301(7):110320. doi: 10.1016/j.jbc.2025.110320. Epub 2025 May 30. J Biol Chem. 2025. PMID: 40451434 Free PMC article.
-
From aging to Alzheimer's disease: concordant brain DNA methylation changes in late life.medRxiv [Preprint]. 2025 Jun 18:2025.06.17.25329345. doi: 10.1101/2025.06.17.25329345. medRxiv. 2025. PMID: 40585142 Free PMC article. Preprint.
-
Exploring Epigenetic Complexity in Regulation of Hematopoietic Stem Cells Niche: A Mechanistic Journey from Normal to Malignant Hematopoiesis.Adv Exp Med Biol. 2025;1483:49-67. doi: 10.1007/5584_2024_846. Adv Exp Med Biol. 2025. PMID: 39841383 Review.
-
Deciphering the role of Wnt signaling in acute myeloid leukemia prognosis: how alterations in DNA methylation come into play in patients' prognosis.J Cancer Res Clin Oncol. 2020 Dec;146(12):3097-3109. doi: 10.1007/s00432-020-03407-3. Epub 2020 Sep 27. J Cancer Res Clin Oncol. 2020. PMID: 32980885 Free PMC article. Review.
References
-
- Swerdlow SH et al. (Lyon: IARC, 2008).
-
- Eriksson A, Lennartsson A, Lehmann S. Epigenetic aberrations in acute myeloid leukemia: early key events during leukemogenesis. Exp Hematol. 2015;43:609–24. 10.1016/j.exphem.2015.05.009. https://doi.org:. - PubMed
-
- Mehdipour P, Santoro F, Minucci S. Epigenetic alterations in acute myeloid leukemias. FEBS J. 2015;282:1786–800. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
